Hormone replacement in patients with hypopituitarism: a practical approach
Hypopituitarism affects about one in 2000 people and requires long-term, often lifelong, management. Implementing a practical, stepwise approach to hormone assessment and replacement is important for all clinicians and can prevent life-threatening complications. This includes an understanding of the sequence and method of central adrenal insufficiency, central hypothyroidism and hypogonadism treatment, but also extends more broadly to cardiovascular and bone health.
- Hypopituitarism is characterised by the inadequate secretion of pituitary hormones.
- The sequence of anterior pituitary hormone replacement is an important consideration. Cortisol should be replaced first, followed by thyroxine, sex hormones and, lastly, growth hormone. This avoids the onset of a life-threatening adrenal crisis and incorrect interpretation of hormone studies based on other untreated deficiencies.
- Corticosteroid replacement in pituitary disease should not be discontinued without endocrinologist input. On sick days, corticosteroid doses should be doubled or tripled depending on the degree of symptom severity.
- Thyroid-stimulating hormone evaluation is not helpful to guide thyroxine replacement; instead, the treatment goal is a free thyroxine level in the upper half of the normal range.
- There is no evidence that testosterone replacement to physiological ranges increases the risk of prostate cancer.
- Symptoms of growth hormone deficiency can be subtle. Some patients will not experience symptom improvement with treatment.
- Patients with arginine vasopressin deficiency receiving desmopressin replacement should drink to thirst to maintain normal sodium levels. The lowest dose should be prescribed to control polyuria or polydipsia.
Pituitary hypofunction affects about one in 2000 people, and its incidence is on the rise, at least partly due to the increasing use of cancer immunotherapy, which frequently results in hypophysitis.1-4 Treatment of hypopituitarism can span decades and is often lifelong. It is therefore valuable for primary care physicians to have a practical approach to hormone evaluation and appropriate replacement. This article provides an overview of the pathophysiology of hypopituitarism, with an emphasis on the importance of appropriate hormone evaluation and replacement therapy.
Pituitary function and hypopituitarism
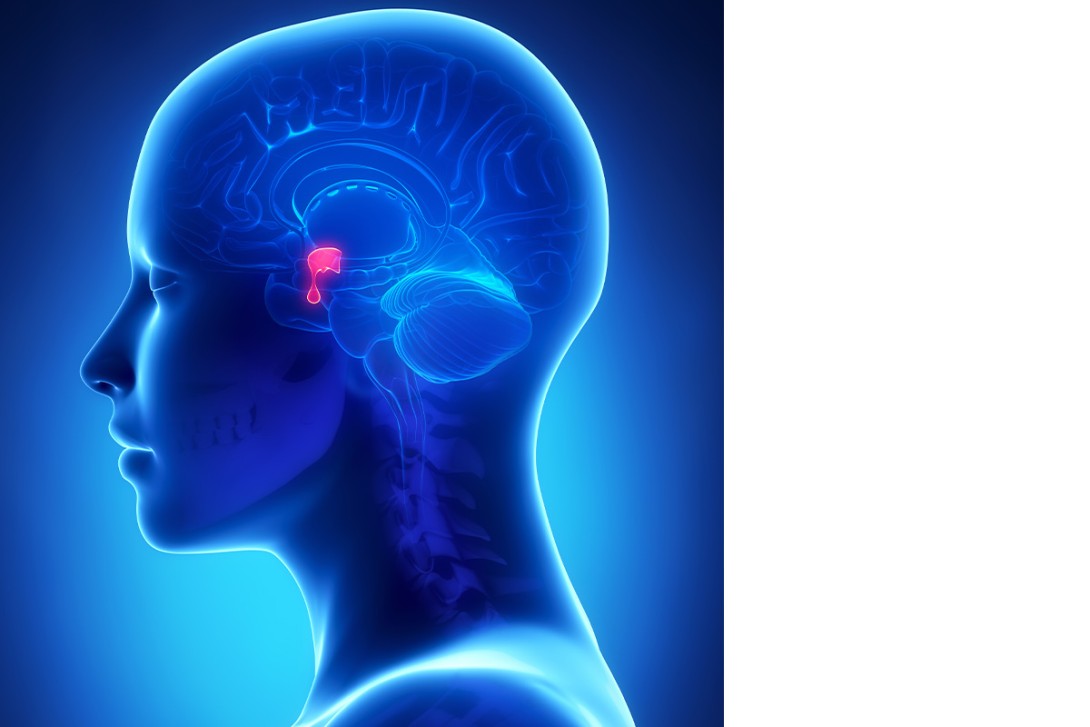
The pituitary is often referred to as the master gland, mostly acting as an intermediary between the hypothalamus and end-organ glands (primary glands), which produce the hormones that are active in body tissues. The anterior pituitary (adenohypophysis) produces six hormones: adrenocorticotropin hormone (ACTH), thyroid-stimulating hormone (TSH), follicle-stimulating hormone (FSH), luteinising hormone (LH), growth hormone (GH) and prolactin. When an anterior pituitary hormone deficiency causes deficiency of end-organ hormone production, the condition is considered ‘secondary’ or ‘central’.5 The posterior pituitary (neurohypophysis) releases two hormones that are synthesised in the hypothalamus: arginine vasopressin (AVP) and oxytocin (Figure).

Hypopituitarism arises because of pituitary or hypothalamic disease, such as pituitary, suprasellar or parasellar tumours; surgery; irradiation; traumatic brain injury; infiltrative disease; vascular insult; or drug effects (Box 1). Hypopituitarism is associated with increased mortality.6,7 Loss of hormonal function can be singular but is often progressive depending on the aetiology. When managing the condition, it is important that hormones be replaced in a certain sequence to avoid potentially life-threatening consequences. A full assessment of pituitary function is therefore necessary in all patients with a suspected or confirmed pituitary hormone abnormality.7 Changes in one type of hormone replacement may affect the other axes, necessitating an integrated approach to replacement regimens. The significance of low prolactin and oxytocin levels is currently unclear, and there is no role for replacement of these hormones.

Hypothalamic–pituitary–adrenal axis
Cortisol, a glucocorticoid stress hormone, is essential in human physiology, and its deficiency is life threatening. The lowest tolerated dose of glucocorticoid replacement is recommended to mimic physiological patterns and avoid detrimental effects of overreplacement, while minimising the risk of inadequate replacement in times of stress, which may otherwise result in an adrenal crisis.8 Central adrenal insufficiency, characterised by low ACTH levels and resulting in hypocortisolism, is present in up to one-third of patients with hypopituitarism. It differs from primary adrenal insufficiency (e.g. Addison’s disease) in that, typically, only glucocorticoid (and not mineralocorticoid) replacement is required.6 This is because ACTH primarily controls cortisol production, whereas aldosterone production is also regulated by the renin–angiotensin system.
When to suspect central adrenal insufficiency
Features suggestive of adrenal insufficiency include fatigue, nausea, weight loss and postural hypotension. Cortisol levels less than 100 nmol/L, measured at 8.00 to 9.00 am, are convincing for the presence of hypocortisolism, whereas intermediate levels are suggestive and should precede confirmatory testing with a short Synacthen (tetracosactide acetate) test.8 A borderline level in our local practice is a morning cortisol level of 100 to 250 nmol/L, but there is considerable variation between assays and so the upper threshold may be as high as 360 nmol/L, depending on local protocols.9 A short Synacthen test can be conducted to assess the adrenal response to a large dose (250 mcg) of synthetic ACTH over 60 minutes. This test relies on the presence of adrenal atrophy, which is a feature of chronic ACTH deficiency. Short Synacthen testing for central hypocortisolism should therefore only be performed after a six-week lag from hypothalamic–pituitary–adrenal insult. A typical cortisol response exceeds 400 to 450 nmol/L.8,10
It is crucial to ensure the correct timing of cortisol tests and adhere to local reference ranges, as there is considerable diurnal and interassay variation; older-generation cortisol assays typically yield high readings.9,11 The diagnosis of central, rather than primary, adrenal insufficiency is established by subsequently demonstrating a low or inappropriately normal ACTH level (the sample of which must be collected on ice).8
Managing hypocortisolism in pituitary disease
Hydrocortisone is the preferred glucocorticoid for replacement, as it is bioidentical to endogenous cortisol.12 The leading alternatives are cortisone acetate, which also allows for variable dosing to mimic normal endogenous variations, and prednisolone, which does not allow for diurnal variations but may aid compliance due to its single daily dosing (Table).

The typical dose of hydrocortisone in adults with hypocortisolism is 15 to 20 mg daily.6 Body weight is the most important predictor of hydrocortisone clearance in adults.13 A weight-based calculation may be used, with evidence that a dose in the range of 0.20 to 0.25 mg/kg body weight is associated with the lowest cardiometabolic risk.14 It is helpful to note that hydrocortisone is available in 20 mg and 4 mg tablets, and that cortisol follows a diurnal rhythm with a peak on waking and a nadir overnight.12 Hence, a typical regimen for an adult with a body weight of 70 kg is 10 mg in the morning and 4 to 8 mg in the early afternoon, or 10 mg in the morning, 4 mg in the early afternoon and 4 mg in the early evening.
Stress dosing of corticosteroids
Hypocortisolism can be fatal, particularly during times of increased physiological stress when the cortisol demand is increased.15 All patients with adrenal insufficiency, regardless of the aetiology, should have a written sick day management plan and carry a medical alert bracelet (or equivalent). Ideally, a close contact will have been trained in parenteral hydrocortisone administration, and patients should always have an in-date kit on hand. Patients are instructed to double or triple their corticosteroid dose for two to three days, depending on the degree of illness. If they are unable to take oral tablets or if the illness progresses, urgent parenteral corticosteroid administration and hospital admission is required.
Increased demand for cortisol is also observed in the perioperative period. A dose of 50 mg intravenous hydrocortisone should be given at the time of anaesthetic induction or at procedure commencement (if no anaesthetic is given). Postoperative requirements depend on the degree of surgery. Information on precise dosing is available from Hormones Australia (https://www.hormones-australia.org.au/patient-resources/).
Hypothalamic–pituitary–thyroid axis
Central hypothyroidism is caused by inadequate TSH signalling to the thyroid. This is demonstrated by a notably low or inappropriately normal TSH level with low thyroid hormone levels. If the results do not correspond to the clinical picture or are obtained after a recent illness, sick euthyroid syndrome should be considered, and measurements of TSH, free thyroxine and free triiodothyronine levels should be repeated.16
Management of central hypothyroidism
Adrenal insufficiency must be excluded before starting thyroid hormone replacement. Starting thyroxine in the setting of untreated adrenal insufficiency can accelerate the metabolism of the low residual cortisol and precipitate adrenal crises.6
Thyroid hormone replacement is similar to hormone replacement for primary hypothyroidism (e.g. associated with Hashimoto’s thyroiditis), using an estimated dose of 1.6 mcg/kg/day of levothyroxine. Patients taking levothyroxine are usually advised to take their tablets at least 30 minutes before or after taking other tablets and other oral intake (apart from water). However, this is often impractical in the setting of panhypopituitarism, as patients require hydrocortisone on waking for symptomatic relief. In such cases, thyroxine and hydrocortisone should be consistently taken together and at least 30 minutes before or after other oral intake. The dose of thyroxine can then be titrated according to the results of repeated blood tests, accepting that the dose requirement may be higher to compensate for theoretically reduced absorption.
Monitoring of central hypothyroidism
The major difference between primary and central hypothyroidism is in monitoring. By definition, TSH levels are inadequate in central hypothyroidism and are therefore not useful to monitor.16 A low TSH level should not be confused with levothyroxine over-replacement. Free thyroxine should be used for titration, aiming for a target level in the mid to upper half of the normal range.6
Hypothalamic–pituitary–gonadal axis
Hypogonadism typically manifests as reduced energy, muscle strength and libido in men; menstrual irregularities, or oligomenorrhoea or amenorrhoea in women; and subfertility or infertility, and low bone density in either sex.6 Central hypogonadism should be suspected if low sex hormone levels are accompanied by inappropriately normal or low FSH and LH levels.17 Functional hypogonadism should be excluded if a clear pituitary disorder is not present.18 This may result from acute or critical illness, anorexia nervosa, obesity (in men), obstructive sleep apnoea or the use of certain medications (e.g. opioids).
A note on hyperprolactinaemia
Hyperprolactinaemia should be excluded as a cause of central hypogonadism.18 If detected, the cause of hyperprolactinaemia should be investigated, and the role of treatment (e.g. antipsychotic medication withdrawal, dopamine agonist therapy or surgical resection) should be considered after endocrinological review. If irregular prolactin levels are the primary cause, normalisation will usually restore the hypothalamic–pituitary–gonadal axis. If hypogonadism persists despite prolactin normalisation, testosterone or oestrogen/progesterone replacement may be required.
Management of central hypogonadism in men
In Australia, oral, transdermal (creams, gels and patches) and intramuscular injection testosterone preparations are available. The choice of therapy is based on individual requirements.18 GPs can prescribe testosterone via the PBS, after endorsement by a treating endocrinologist, urologist or sexual health physician (Fellow of the Australasian Chapter of Sexual Health Medicine).
The monitoring of patients on testosterone therapy is guided by routine clinical review with thorough history taking and examination.18 Routine assessments of serum testosterone levels with sex hormone-binding globulin and haematocrit is recommended.
Bone densitometry, sleep study and prostate-specific antigen (PSA) level monitoring may be tailored to individual patient needs after baseline screening.5,6 Restoration of physiological testosterone levels is not thought to cause prostate cancer; however, existing prostate cancer can progress with testosterone exposure. Hence, PSA levels should be measured at baseline before starting testosterone therapy to rule out high PSA levels requiring urological evaluation. However, the decision to pursue ongoing PSA monitoring should be based on the same considerations as in the general population.18
Measurements of serum testosterone levels in patients taking daily treatments should be acquired two to three hours after their usual dose. Three-monthly monitoring of intramuscular testosterone levels is performed the week before the next scheduled dose (trough level), but this does not delay the dose. Therapeutic targets incorporate both patients’ physical needs and achieving testosterone levels in the middle of the normal range.18 An elevated haematocrit level (≥0.54%) should prompt a reduction in testosterone dosage. Other strategies for managing polycythaemia include switching from intramuscular to transdermal testosterone preparations and, ultimately, venesection.
Management of central hypogonadism in women
Female hypogonadism may be associated with premature cardiovascular disease, reduced BMD and reduced quality of life.6,7 Current recommendations include the use of sex hormone replacement until the natural age of menopause.
In the hypopituitary setting, transdermal oestrogen replacement is preferred, as first-pass metabolism of oral oestrogen attenuates GH and insulin-like growth factor-1 (IGF-1) action and metabolism.19 Furthermore, transdermal oestrogen has been shown to yield greater gains in BMD than oral oestrogen in central hypogonadism.20 If oral oestrogen is used, higher dose requirements for GH should be anticipated.6 The Australasian Menopause Society provides a discussion of oestrogen and progesterone replacement options (https://www.menopause.org.au/hp/management). Evidence does not currently support the routine use of androgen therapy (testosterone or dehydroepiandrosterone) in women.21,22
Monitoring following the initiation of replacement therapy is primarily symptom based. BMD should be measured periodically if it is low at baseline (e.g. biannually, or 12 months after the treatment changes).
Fertility in hypopituitarism
Both male and female fertility are impaired in central hypogonadism. Assisted reproductive treatments (i.e. ovulation induction or in vitro fertilisation) are often required for conception, with pregnancy rates ranging from 47 to 76% in small observational studies.17,21 Women with central hypogonadism who wish to conceive should be offered ovulation induction with gonadotropins. In men with central hypogonadism who wish to conceive, testosterone replacement should be paused, as this directly inhibits spermatogenesis, and human chorionic gonadotropin with or without FSH should be used instead to induce spermatogenesis. Several months of treatment may be required to induce adequate sperm counts.23,24
Growth hormone
Adult-onset GH deficiency can be clinically subtle but impact metabolic health and cardiovascular risk.7 Because of interactions between the various pituitary hormones and given that GH is the least critical hormone to replace, the other pituitary axes should be investigated and any abnormalities treated before GH is assessed.25
IGF-1 levels may be used to screen for GH deficiency; a definitive diagnosis is made based on the results of an insulin tolerance test or glucagon stimulation test, with assessment of the peak GH level.6 GH replacement consists of daily subcutaneous injections of recombinant human GH. Weekly preparations exist but are not yet available via the PBS.26 Recombinant human GH is not recommended in patients with active malignancy or during pregnancy.25 The GH dose is titrated to symptoms of GH deficiency or excess and the IGF-1 level, which should be mid-normal.
During pregnancy, placental GH production mitigates the need for exogenous GH replacement. Although GH replacement is typically ceased on confirmation of pregnancy, some experts advocate for weaning after the first trimester.21,27 Limited data suggest that GH continuation in pregnancy is not harmful, but prospective studies are needed.28
Arginine vasopressin
AVP, also known as antidiuretic hormone, is released from the posterior pituitary in response to increased serum osmolality and acts on the renal tubules to retain water. AVP deficiency (AVP-D), previously termed central diabetes insipidus, may be complete or partial, causing renal water wasting and significant polyuria.29
Most patients with AVP-D retain their sense of thirst, which is important for salt and water balance. Provided patients are able to drink to thirst to replace urinary losses, AVP-D management is straightforward.30 Adipsic AVP-D is rare, being found only with extensive sellar masses or injuries that involve the hypothalamic regions responsible for thirst.
Initial investigations for polyuria-polydipsia syndrome include measurements of 24-hour urine volume and osmolality with paired serum osmolality, sodium, glucose, calcium and copeptin. Confirmatory testing (e.g. via the hypertonic saline-stimulated copeptin test) may be required to differentiate AVP-D from primary polydipsia.29,31,32
The treatment of AVP-D involves the use of desmopressin, which is a synthetic analogue of vasopressin with a longer half-life. The aim of treatment is to provide relief from the excessive urination and thirst, which otherwise interfere with daily activities and sleep. Dosing is titrated to relieve symptoms while avoiding excessive water retention leading to hyponatraemia. Overtreatment with desmopressin can cause dangerous hyponatraemia, hence, erring on the side of lower desmopressin doses and drinking to thirst is typically the safest option.29 Withholding desmopressin periodically until breakthrough polyuria develops has been shown to lower rates of hospitalisation for hyponatraemia.32-34
Sick day management of AVP-D
Sick days can generally be managed using the usual rule of drinking to thirst and aiming for equal fluid input and output. This can be difficult with gastrointestinal illnesses, such as diarrhoea, and inpatient management may be required under the care of an endocrinologist.35
Broader considerations in hypopituitarism
Patients with hypopituitarism have an increased cardiovascular risk. Modifiable cardiovascular risk factors should therefore be considered routinely and optimised where possible, including smoking cessation and lipid assessment.6
Bone density is negatively affected by hypogonadism, GH deficiency and supraphysiological replacement of cortisol and thyroxine, but typically improves, or at least stabilises, with appropriate hormonal replacement, adequate calcium and vitamin D consumption and engaging in weightbearing exercise.6 Bone densitometry should be considered in these settings.
Conclusion
Hypopituitarism affects a broad range of physiological systems and necessitates stepwise investigation and management. Identification and treatment of central adrenal insufficiency prior to starting other therapies is key, as is recognising the differences between primary and central hormone deficiencies. Box 2 lists some resources for patients and GPs. ET

COMPETING INTERESTS: None.
References
1. Regal M, Páramo C, Sierra SM, et al. Prevalence and incidence of hypopituitarism in an adult Caucasian population in northwestern Spain. Clin Endocrinol (Oxf) 2001; 55: 735-740.
2. Fernandez-Rodriguez E, Lopez-Raton M, Andujar P, et al. Epidemiology, mortality rate and survival in a homogeneous population of hypopituitary patients. Clin Endocrinol (Oxf) 2013; 78: 278-284.
3. Rushworth RL, Torpy DJ. The changing epidemiology of adrenal insufficiency: iatrogenic factors predominate. J Endocr Soc 2023; 7: bvad017.
4. Raschi E, Fusaroli M, Massari F, et al. The changing face of drug-induced adrenal insufficiency in the Food and Drug Administration adverse event reporting system. J Clin Endocrinol Metab 2022; 107: e3107-e3114.
5. Alexandraki K, Grossman A. Management of hypopituitarism. J Clin Med 2019; 8: 2153.
6. Fleseriu M, Hashim IA, Karavitaki N, et al. Hormonal replacement in hypopituitarism in adults: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2016; 101: 3888-3921.
7. Pappachan JM, Raskauskiene D, Kutty VR, et al. Excess mortality associated with hypopituitarism in adults: a meta-analysis of observational studies. J Clin Endocrinol Metab 2015; 100: 1405-1411.
8. Grossman AB. The diagnosis and management of central hypoadrenalism. J Clin Endocrinol Metab 2010; 95: 4855-4863.
9. Sbardella E, Isidori AM, Woods CP, et al. Baseline morning cortisol level as a predictor of pituitary-adrenal reserve: a comparison across three assays. Clin Endocrinol 2017; 86: 177-184.
10. Kazlauskaite R, Evans AT, Villabona CV, et al. Corticotropin tests for hypothalamic-pituitary- adrenal insufficiency: a metaanalysis. J Clin Endocrinol Metab 2008; 93: 4245-4253.
11. Brown S, Hadlow N, Badshah I, et al. A time-adjusted cortisol cut-off can reduce referral rate for Synacthen stimulation test whilst maintaining diagnostic performance. Clin Endocrinol 2017; 87: 418-424.
12. Isidori AM, Arnaldi G, Boscaro M, et al. Towards the tailoring of glucocorticoid replacement in adrenal insufficiency: the Italian Society of Endocrinology Expert Opinion. J Endocrinol Investig 2020; 43: 683-696.
13. Mah PM, Jenkins RC, Rostami-Hodjegan A, et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol (Oxf) 2004; 61: 367-375.
14. Zueger T, Kirchner P, Herren C, et al. Glucocorticoid replacement and mortality in patients with nonfunctioning pituitary adenoma. J Clin Endocrinol Metab 2012; 97: E1938-E1942.
15. Rushworth RL, Torpy DJ, Falhammar H. Adrenal crisis. NEJM 2019; 381: 852-861.
16. Lania A, Persani L, Beck-Peccoz P. Central hypothyroidism. Pituitary 2008; 11: 181-186.
17. Garmes HM, Boguszewski CL, Miranda PAC, et al. Management of hypopituitarism: a perspective from the Brazilian Society of Endocrinology and Metabolism. Arch Endocrinol Metab 2021; 65: 212-230.
18. Bhasin S, Brito JP, Cunningham GR, et al. Testosterone therapy in men with hypogonadism: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2018; 103: 1715-1744.
19. Shoung N, Ho KKY. Managing estrogen therapy in the pituitary patient. J Endocr Soc 2023; 7: bvad051.
20. Ackerman KE, Singhal V, Baskaran C, et al. Oestrogen replacement improves bone mineral density in oligo-amenorrhoeic athletes: a randomised clinical trial. Br J Sports Med 2019; 53: 229-236.
21. Vila G, Fleseriu M. Fertility and pregnancy in women with hypopituitarism: a systematic literature review. J Clin Endocrinol Metab 2020; 105: dgz112.
22. Zang H, Davis SR. Androgen replacement therapy in androgen-deficient women with hypopituitarism. Drugs 2008; 68: 2085-2093.
23. Marques JVO, Boguszewski CL. Fertility issues in aggressive pituitary tumors. Rev Endocr Metab Disord. 2020; 21: 225-233.
24. Andreassen M, Juul A, Feldt-Rasmussen U, et al. Semen quality in patients with pituitary disease and adult-onset hypogonadotropic hypogonadism. Endocr Connect 2018; 7: 523-533.
25. Molitch ME, Clemmons DR, Malozowski S, et al. Evaluation and treatment of adult growth hormone deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2011; 96: 1587-1609.
26. Chatelain P, Malievskiy O, Radziuk K, et al. A randomized phase 2 study of long-acting TransCon GH vs daily GH in childhood GH deficiency. J Clin Endocrinol Metab 2017; 102: 1673-1682.
27. Vila G, Luger A. Growth hormone deficiency and pregnancy: any role for substitution? Minerva Endocrinol 2018; 43: 451-457.
28. Biller BMK, Höybye C, Carroll P, et al. Pregnancy outcomes in women receiving growth hormone replacement therapy enrolled in the NordiNet® International Outcome Study (IOS) and the American Norditropin® Studies: Web-Enabled Research (ANSWER) Program. Pituitary 2021; 24: 611-621.
29. Christ‐Crain M, Winzeler B, Refardt J. Diagnosis and management of diabetes insipidus for the internist: an update. J Intern Med 2021; 290: 73-87.
30. Christ-Crain M, Bichet DG, Fenske WK, et al. Diabetes insipidus. Nat Rev Dis Primers 2019; 5: 54.
31. Christ-Crain M, Fenske WK. Copeptin in the differential diagnosis of hypotonic polyuria. J Endocrinol Investig 2020; 43: 21-30.
32. Tomkins M, Lawless S, Martin-Grace J, et al. Diagnosis and management of central diabetes insipidus in adults. J Clin Endocrinol Metab 2022; 107: 2701-2715.
33. Behan LA, Sherlock M, Moyles P, et al. Abnormal plasma sodium concentrations in patients treated with desmopressin for cranial diabetes insipidus: results of a long-term retrospective study. Eur J Endocrinol 2015; 172: 243-250.
34. Atila C, Loughrey PB, Garrahy A, et al. Central diabetes insipidus from a patient’s perspective: management, psychological co-morbidities, and renaming of the condition: results from an international web-based survey. Lancet Diabetes Endocrinol 2022; 10: 700-709.
35. Baldeweg SE, Ball S, Brooke A, et al. Society for Endocrinology clinical guidance: inpatient management of cranial diabetes insipidus. Endocr Connect 2018; 7: G8-G11.
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.