The many faces of phaeochromocytoma
Adrenal gland disorders
Endocrine diseases
Phaeochromocytomas are rare catecholamine-secreting tumours that pose significant risk of cardiovascular morbidity and mortality and potential for metastasis. Delayed or missed diagnosis is common due to their wide spectrum of clinical manifestations.
- Timely diagnosis of a phaeochromocytoma is important because of the associated cardiovascular morbidity and mortality and potential for metastatic disease.
- Clinical presentation is highly variable and features may be nonspecific, mimicking a spectrum of medical and psychological conditions.
- The most common features of a phaeochromocytoma are hypertension, headache, sweating, palpitations and anxiety.
- Patients may be asymptomatic, especially with the rising detection of incidental adrenal lesions.
- Diagnosis requires biochemical assessment for catecholamine excess, followed by imaging.
- Of people with phaeochromocytomas or paragangliomas, 40% carry an autosomal dominant germline mutation. Genetic testing should be considered for all patients.
Phaeochromocytomas and paragangliomas are rare catecholamine-secreting tumours. Phaeochromocytomas arise from the adrenal medulla whereas sympathetic paragangliomas arise from the sympathetic and parasympathetic ganglia. These tumours have an estimated incidence of 0.6 to 0.8 per 100,000 person-years, but this is likely to be an underestimate due to missed diagnoses, with a higher frequency noted in autopsies.1-3 Phaeochromocytomas make up about 85% of these tumours,4 and are the main focus of this article.
The diagnosis of phaeochromocytoma can be delayed or missed due to the wide spectrum of potential clinical presentations. Timely diagnosis is paramount because of the association of phaeochromocytoma with potentially catastrophic cardiovascular morbidity, risk of metastatic disease (which occurs in 10 to 15% of patients with phaeochromocytomas or paragangliomas) and potential for detection of a germline mutation.3,5
Presentation
The clinical manifestations of phaeochromocytomas are attributable to the actions of excess circulating catecholamines on the sympathetic nervous system. Symptoms and signs are thus nonspecific. Presentation at diagnosis is highly variable, ranging from catecholaminergic crisis with multiorgan failure, asymptomatic hypertension, normotension or hypotension to mimicking of psychological conditions.
The often described classic triad comprises paroxysmal headache, spontaneous or excessive sweating and palpitations. However, only a minority of patients present with this classic triad.
The most common feature of phaeochromocytoma is hypertension, which may be paroxysmal or sustained. Postural hypotension may also occur. Other cardiovascular manifestations include chest pain and nonspecific electrocardiographic changes that may mimic ischaemia. Presenting features may also include pallor, anxiety or panic attacks, tremor, fatigue, gastrointestinal manifestations and hyperglycaemia.3,6 Rarely, patients may present with catecholaminergic crisis causing organ dysfunction (as illustrated in the case in Box 1). Other features include haemodynamic instability with severe hypertension or hypotension, arrhythmia, myocardial infarction, cardiomyopathy, encephalopathy, stroke and fever.7
{kind=link}
Earlier diagnosis is increasingly being made in relatively asymptomatic patients due to the detection of incidental adrenal masses or through germline mutation testing. Phaeochromocytomas comprise 5% of adrenal incidentalomas.2
Given the wide spectrum of clinical manifestations and potential for asymptomatic disease, it is important to consider the diagnosis of phaeochromocytoma if consistent features, including atypical presentations, are present (as illustrated in the case described in Box 2). A biochemical assessment should also be conducted in patients with adrenal masses, including those who are asymptomatic.
{kind=link}
Biochemical diagnosis and imaging
Indications for biochemical assessment for catecholamine excess are a clinical suspicion of phaeochromocytoma or paraganglioma, detection of an adrenal or retroperitoneal mass, and carriers of phaeochromocytoma or paraganglioma susceptibility genes.3
Initial biochemical assessment should comprise measurement of fasting plasma free metanephrines (this includes metanephrine [also called metadrenaline] and normetanephrine [also called normetadrenaline]) and 3-methoxytyramine levels, or alternatively measurement of 24-hour urinary metanephrine levels. These catecholamine metabolites have a sensitivity of 97% and have greater diagnostic accuracy than measurement of catecholamines (adrenaline, noradrenaline and dopamine).2 Measurement of fasting plasma 3-methoxytyramine levels is carried out to increase sensitivity and for detection of rare tumours that predominantly secrete dopamine, particularly if paraganglioma or metastatic disease are suspected.
Biochemical investigations are prone to false positives. These investigations should be performed in the fasting state, in supine position and avoided at times of stress because of stimulation of the sympathetic nervous system. Metanephrine levels more than three times the upper reference limit are rarely false positive.8 Medications that may interfere with biochemical testing should be tapered and withheld for at least two weeks before testing (Box 3).9-11 For example, tricyclic antidepressants and nonselective alpha blockers such as phenoxybenzamine increase plasma and urinary normetanephrine levels, whereas beta adrenoceptor blockers may cause false elevation of plasma and urinary metanephrine levels.9 Marginal elevation of screening test results warrants consideration of factors causing false elevation and repeat of the investigations after eliminating these factors if possible.
{kind=link}
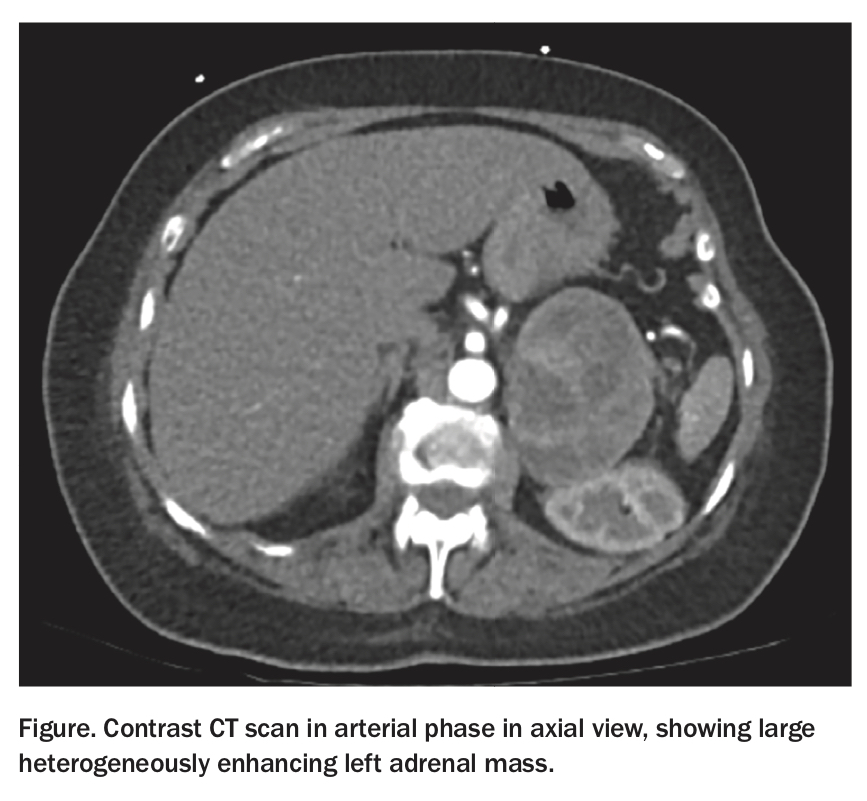
Imaging for localisation should generally be performed only after demonstrating biochemical evidence of catecholamine excess. Abdominal CT with contrast is preferred and is safe as there is no risk of exacerbation of hypertension.12 MRI may be performed if CT with contrast is contraindicated or to assess for extra-adrenal disease if CT is unremarkable. Bilateral disease and extra-adrenal disease are associated with a higher risk of an underlying germline mutation. Phaeochromocytomas typically show marked and heterogeneous hyperenhancement on CT (Figure), and peripheral rim enhancement is characteristic of a phaeochromocytoma with central necrosis. In contrast to lipid-rich adenomas, the unenhanced attenuation is almost always greater than 10 Hounsfield units, with less than 50% contrast washout at 10 minutes. There is, however, considerable variability in radiological appearance.13
{kind=link}
Functional imaging may be considered following CT and/or MRI. It is generally considered to have additional benefit only in specific circumstances, such as if there is known metastasis or a high risk of metastatic disease (e.g. in patients with certain underlying genetic mutations). Traditionally, metaiodobenzylguanidine scintigraphy (MIBG) has been used for localisation, if CT and/or MRI are negative, and for detection of metastatic disease. However, sensitivity for detection of metastases or paraganglioma may be higher with fluorodeoxyglucose positron emission tomography and 68-gallium DOTATE positron emission tomography, depending on the underlying genetic mutation, if present, location of the primary tumour and location of metastases.14,15
Management
Patients with suspected or diagnosed phaeochromocytoma should be referred to an endocrinology service to guide management. Surgical resection is the cornerstone of therapy for phaeochromocytoma, but appropriate preoperative evaluation and medical preparation is crucial to minimise the risk of perioperative cardiovascular complications from the surge of catecholamine release in the setting of operative manipulation. Perioperative mortality is low with suitable medical preparation.3
Patients should be started on an alpha-adrenergic receptor blocking agent one to two weeks before scheduled surgery. It is crucial that adequate alpha blockade is achieved before beta adrenergic receptor blocking agents are introduced, with monitoring of sitting and upright blood pressures to guide dosing. Beta blockade should not be started before this due to the risk of unopposed alpha adrenergic stimulation precipitating a hypertensive crisis.
Phenoxybenzamine, a nonselective alpha adrenergic blocker, is typically used starting at 10 mg twice a day and the dose is increased until a target blood pressure of 120/80 mmHg with postural drop of up to 20/10 mmHg is achieved. Selective alpha blockers such as prazosin can also be used. After adequate alpha blockade is achieved, a beta blocker, such as atenolol or metoprolol, is subsequently administered to control tachycardia, with a target heart rate of 60 beats per minute.2 Patients should be cautioned regarding potential adverse effects of medications, including nasal stuffiness and postural symptoms. In patients with no underlying cardiac or renal failure, salt and fluid intake are to be increased to reverse the catecholamine-induced volume contraction. In addition, preoperative intravenous saline may be used to prevent prolonged and severe hypotension postoperatively.
Retroperitoneal laparoscopic adrenalectomy, instead of open surgery, is now often performed by experienced surgeons and is considered standard of care. Postoperative follow up with an endocrinology service is essential to confirm complete tumour resection and for surveillance of recurrent disease.2,5
Genetic testing
Of patients with phaeochromocytomas or paragangliomas, 40% have an underlying germline mutation. Detection of germline mutations is important given the implications for family members and the higher risk of metastatic disease associated with some germline mutations (as illustrated in the case in Box 4).
{kind=link}
Disease-causing germline mutations include RET mutations causing multiple endocrine neoplasia type 2A or 2B, succinate dehydrogenase (SDH) mutations (composed of four subunits – SDHA, SDHB, SDHC, SDHD), succinate dehydrogenase complex assembly factor 2 (SDHAF), von Hippel Lindau (VHL), neurofibromatosis type 1, MYC-associated factor X (MAX) and transmembrane protein 127 (TMEM127), which are associated with variable penetrance of phaeochromocytomas and paragangliomas, risk of metastatic disease and different biochemical phenotypes. For example, SDHB and VHL are associated with predominantly noradrenaline-secreting tumours. SDHB mutations confer higher risk of metastasis.3,16
It is advised that genetic testing be considered for all patients with phaeochromocytomas or paragangliomas, preferably with the assistance of clinical genetic services. Testing is particularly important if suspicious features, such as significant family history, bilateral or metastatic disease or presence of associated syndromal features such as other tumour types, are present. Next generation sequencing, which enables all genes known to be involved in phaeochromocytomas and paragangliomas to be simultaneously evaluated, is currently used mainly for research purposes. It may become widely available as a genetic diagnostic tool for phaeochromocytomas and paragangliomas in the future once it becomes more affordable.
Conclusion
Patients with phaeochromocytomas can present with a wide spectrum of clinical manifestations, most symptoms being nonspecific and mimicking both medical and psychological conditions. Considering the diagnosis in the appropriate clinical setting is important, especially due to the cardiovascular complications of untreated disease. It is also paramount to consider the potential for an underlying germline mutation, which may help to guide early diagnosis and treatment in family members. ET
References
Plasma_Metanephrine_Normetanephrine_Fact_Sheet_Jan_2018_-_01.pdf (accessed April 2020).
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.