Atypical diabetes: clues to causes, conundrums and care
Dr Wong is a Senior Staff Specialist Endocrinologist at Royal Prince Alfred Hospital, Sydney; and a Clinical Associate Professor at The University of Sydney, Sydney, NSW.
Endocrine diseases
Diabetes complications
Occasionally, clinicians see patients with diabetes that does not fit the usual pattern of either type 1 or type 2 diabetes and can be considered ‘atypical diabetes’. Subtypes include monogenic, mitochondrial and ketosis-prone diabetes and latent autoimmune diabetes of adults. GPs need a high index of suspicion for these subtypes as treatment and prognosis may vary from those of type 1 and type 2 diabetes.
- Recognition of patients with atypical diabetes is important because of the therapeutic and prognostic implications of this diagnosis.
- Patients with atypical diabetes have often been misdiagnosed with type 1 or type 2 diabetes and in some cases committed to unnecessary insulin therapy and screening for complications.
- Maturity onset diabetes of the young (MODY) is most commonly caused by a mutation in the gene encoding hepatocyte nuclear factor 1-alpha; initial treatment is low-dose sulfonylurea therapy.
- MODY caused by mutations in the gene encoding gluco- kinase does not require treatment outside of pregnancy.
- Type 1 diabetes diagnosed before 6 months of age should prompt screening for neonatal diabetes genes, which may lead to discontinuation of insulin therapy.
- Ketosis-prone diabetes should be considered in patients presenting with ketoacidosis and a type 2 diabetes phenotype.
- Latent autoimmune diabetes in adults can be considered as slowly progressive type 1 diabetes.
- Maternally inherited diabetes and early deafness are clues to the possibility of mitochondrial diabetes.
- Genetic molecular testing is available, and patients with atypical diabetes should be referred for specialist assessment.
A GP is often the first person to diagnose a patient with diabetes mellitus and to determine its ‘type’. This determination is usually viewed through the binary prism of either type 1 or type 2 diabetes, which is understandable given that type 2 diabetes accounts for more than 90% of cases and type 1 diabetes for a further 5 to 10%. However, occasionally clinicians see patients who do not have the fulminant features of type 1 diabetes but are lean, often young and do not have the hallmarks of type 2 diabetes, insulin resistance or the metabolic syndrome, or have overlapping features of type 1 and type 2 diabetes. These patients are not so easily classified and can be considered to have ‘atypical diabetes’.
It is important to remember that diabetes is a large heterogeneous group of disorders, all characterised by defects in insulin secretion and insulin sensitivity to varying degrees, and that rarer subtypes of diabetes exist.1 With increasing knowledge of these subtypes, it has become important for clinicians to identify them more accurately because they have significant implications for prognosis, treatment and, depending on the molecular subtype, counselling and genetic testing. Genetic molecular testing is available, and patients with atypical diabetes should be referred for specialist assessment. Previously, atypical diabetes may have been misclassified as type 1 or type 2 diabetes.
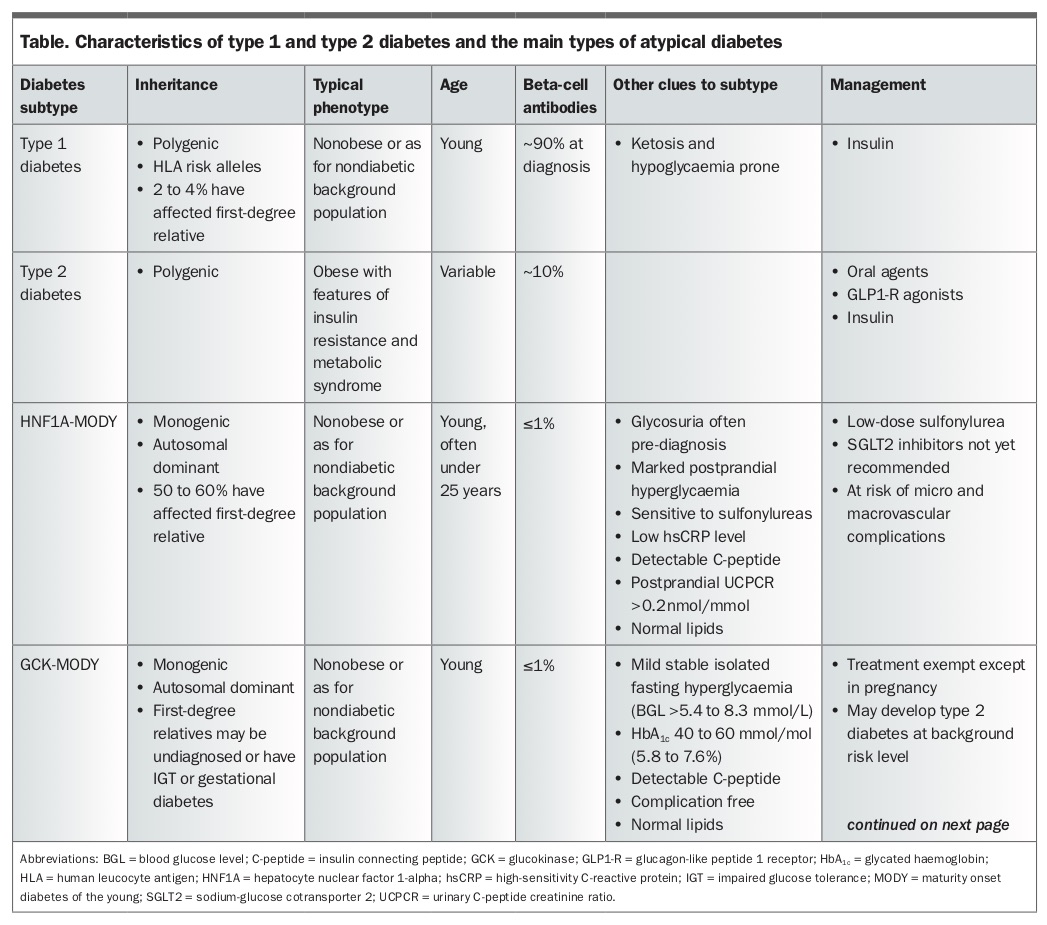
This article provides a practical overview of clinical clues that should alert clinicians to an alternative subtype of diabetes. The implications for patient care are discussed. Subtypes of atypical diabetes described in detail include monogenic diabetes, mitochondrial diabetes, ketosis-prone diabetes and latent autoimmune diabetes of adults (LADA). All these subtypes potentially masquerade as type 1 or type 2 diabetes. Other rarer or medication-induced diabetes subtypes are not discussed. Features of the main atypical diabetes subtypes and type 1 and type 2 diabetes are summarised in the Table Part 1 and Table 1 Part 2.
{kind=link}
{kind=link}
Monogenic diabetes
Monogenic diabetes is a heterogeneous group of disorders that arise from single gene mutations affecting transcription factors and enzymes involved in beta-cell secretory function. Monogenic diabetes can be further generally divided into maturity onset diabetes of the young (MODY) and neonatal diabetes. As monogenic diabetes is often detected in the first two decades of life, patients may be diagnosed with type 1 diabetes and unnecessarily committed to lifelong insulin therapy and the attendant implications. However, patients with monogenic diabetes still have endogenous insulin secretion, and insulin may not be absolutely necessary in their treatment. It is therefore important for clinicians to have a high index of suspicion for monogenic diabetes and to revisit the diagnosis for their patients who have been previously diagnosed with type 1 diabetes.
Maturity onset diabetes of the young
MODY is thought to account for 1 to 2% of all cases of diabetes. Given the overall high prevalence of diabetes, this is a significant number. Important clinical clues to MODY are:
- patients are young at onset
- patients are often lean, although body mass index (BMI) may be a less reliable discriminator as the population BMI increases.
However, unlike patients with typical type 1 diabetes, patients with MODY often have:
- low insulin requirements
- often a stable fasting glucose level
- no beta-cell antibodies (such as insulin autoantibodies and antibodies to glutamic acid decarboxylase, insulinoma-associated autoantigen 2 and zinc transporter)
- no other autoimmunity
- no ketoacidosis
- no signs of insulin resistance or the metabolic syndrome (e.g. acanthosis nigricans, high triglyceride level and low HDL level)
- nonacute presentation.
Other clinical clues come from the family history. MODY is characterised by an autosomal dominant mode of inheritance with a high penetrance (i.e. a high likelihood of disease if the mutation is present), with 50% of offspring affected in multiple generations. Thus, taking a comprehensive family history is important. The presence of multigenerational diabetes (early-onset type 2 or type 1 diabetes) tracking along a single parental line with about 50% of siblings affected should alert the clinician to the possibility of MODY.
At least 14 MODY subtypes have been identified, which generally differ in phenotype, associated conditions and response to therapy. The two most common forms of MODY, accounting for about 80% of cases, are caused by mutations in the genes encoding hepatocyte nuclear factor 1-alpha (HNF1A) or glucokinase (GCK).
HNF1A-maturity onset diabetes of the young
HNF1A-MODY (also known as MODY 3) arises from a mutation in the gene encoding HNF1A, a transcription factor expressed in the liver and pancreas. HNF1A-MODY is the most common form of MODY in Europe, with diabetes typically presenting in adolescence or early adulthood. The glucose pattern is characterised, at least in the early stages, by marked postprandial hyperglycaemia and a fasting glucose level often in the normal range. The two-hour postprandial glucose excursion after an oral glucose tolerance test (OGTT) is often in excess of 5mmol/L. Patients with HNF1A-MODY have increased insulin sensitivity and often significant glycosuria, which may predate the onset of diabetes. They also have underexpression of sodium-glucose cotransporter 2 (SGLT2) in the kidney. It is thought best to avoid SGLT2 inhibitors in the treatment of patients with this subtype of diabetes.
Clinically, patients with HNF1A-MODY have heightened sensitivity to sulfonylureas, and thus a tendency to hypoglycaemia on low-dose sulfonylurea therapy is another diagnostic clue. If patients have a genetic diagnosis confirmed then those using insulin (perhaps having been misdiagnosed with type 1 diabetes) could be trialled on low-dose sulfonylurea therapy and often achieve superior control, although some still progress to requiring insulin therapy with time.
As postprandial hyperglycaemia can be severe, patients with HNF1A-MODY are at risk of complications and an excess risk of cardiovascular disease (CVD) despite normal-to-high HDL levels. Treatment with cholesterol-lowering medications to protect against CVD has been recommended, at least from the age of 40 years.2 Women who have HNF1A-MODY first detected in pregnancy will be diagnosed with gestational diabetes and receive standard management because of the risk of macrosomia.
Another, rarer, form of MODY caused by mutations in the gene encoding hepatocyte nuclear factor 4-alpha (HNF4A) presents with a similar phenotype to HNF1A-MODY. HNF4A-MODY could be considered at the time of genetic testing.
GCK-maturity onset diabetes of the young
GCK is an enzyme involved in insulin secretion, known as the ‘glucose sensor’ of the beta cell. In GCK-MODY (also known as MODY 2), GCK is altered due to a mutation, the net effect being that glucose is regulated normally but at a higher set-point. GCK-MODY is characterised by a mild elevation in fasting glucose level, in the range of more than 5.4 to 8.3mmol/L, without significant postprandial rises so that the OGTT profile is typically flat. The glycated haemoglobin (HbA1c) usually ranges from 40 to 60 mmol/mol (5.8 to 7.6%), with a slight deterioration with age. In contrast to patients with HNF1A-MODY, those with GCK-MODY have fasting hyperglycaemia present at birth, but they are asymptomatic and are thus not identified until incidental testing.
No dietary or pharmacological treatment is necessary for patients with GCK-MODY, with the two caveats discussed below, as studies have found that diabetic complications are not in excess of background.3 In fact, treating the fasting hyperglycaemia in patients with GCK-MODY is difficult, with a poor response to therapy as glucose regulation is normal, albeit at a higher set-point.
There are two caveats to this. Firstly, treatment is offered in pregnancy when the offspring does not carry the mutation and would therefore be at risk of macrosomia if maternal hyperglycaemia is untreated. Fetal genotype may be known from chorionic villous sampling or amniocentesis performed for other reasons. Otherwise, current advice is to observe fetal growth using serial ultrasound examination as a surrogate for fetal genotyping, to guide diabetes management.4
GCK-MODY in pregnant women may be an increasing issue as pregnancy may be the first time that fasting hyperglycaemia is detected, particularly as the fasting diagnostic thresholds for gestational diabetes have been lowered; it has been estimated that 1 to 2% of cases of gestational diabetes are actually GCK-MODY. A BMI less than 25 kg/m2 with a fasting glucose level of 5.5mmol/L or more have been suggested as criteria for genetic screening for GCK-MODY in pregnancy, although these criteria are likely to be less discriminatory in Asian populations.5
The second caveat is that the diagnosis of GCK-MODY does not preclude a risk of developing type 2 diabetes in later life. Arguments have been made to treat patients with GCK-MODY if HbA1c increases repeatedly above 60 mmol/mol (7.6%). However, this approach is debated as it is not known whether it attenuates any excess risk.6
Neonatal diabetes
It is now recognised that diabetes presenting in the neonatal period is due to genetic causes rather than autoimmunity, especially when diagnosed in infants younger than 6 months. Neonatal diabetes may be transient or permanent. Permanent neonatal diabetes is most commonly caused by mutations affecting the ATP-sensitive potassium channel involved in insulin secretion (ABCC8 and KCNJ11 genes encoding the SUR1 and Kir6.2 subunits of the potassium channel).
Current guidelines suggest that patients diagnosed with neonatal diabetes should be immediately referred for genetic testing. However, as mentioned previously, patients may have been misdiagnosed with type 1 diabetes in the past. In some of these, switching from insulin to oral sulfonylurea treatment can improve control and benefit the neurocognitive deficits associated with neonatal diabetes, depending on the mutation present. Although these cases are rare, clinicians should be alert to the possibility in patients who were diagnosed with type 1 diabetes presenting before the age of 6 months.
Diagnosis of monogenic diabetes
In the presence of the clinical phenotype and clues mentioned above, another finding that is helpful for the diagnosis of monogenic diabetes is the absence of beta-cell antibodies. If patients are treated with insulin then a postprandial urinary insulin connecting (C-) peptide to creatinine ratio more than 0.2nmol/mmol differentiates HNF1A- and HNF4A-MODY from type 1 diabetes with high specificity and sensitivity.7 A low level of high-sensitivity C-reactive protein has also been suggested as a pointer to HNF1A-MODY.8
A MODY probability calculator is available online to aid clinical decisions regarding genetic testing in people of European Caucasian descent (www.diabetesgenes.org/mody-probability-calculator). However, this calculator needs validation in other populations. Definitive genetic testing is available from several laboratories. Consultation with specialist endocrine or genetic services is advisable. Genetic testing for neonatal diabetes is currently provided free of charge through funding from the Wellcome Trust.
Ketosis-prone diabetes
Ketosis-prone diabetes is a heterogeneous group aetiologically and probably increasing in prevalence. It is characterised by patient presentation with often unprovoked ketoacidosis in the absence of an autoimmune type 1 diabetes phenotype. The clinical course differs from that of type 1 diabetes as periods of insulin independence are interspersed with periods of acute insulin deficiency and ketoacidosis. Typically, patients with ketosis-prone diabetes have a type 2 diabetes phenotype and are mostly men. Ketosis-prone diabetes is most often seen in non-Caucasian groups, such as Afro-Caribbean and Hispanic peoples, but it has been reported in many ethnicities. This type of diabetes was previously described as ketosis-prone type 2 diabetes or Flatbush diabetes.
Insulin should always be the initial therapy for patients with ketosis-prone type 2 diabetes, but some may be managed with noninsulin treatments with close clinical follow up. Tests for the presence of type 1 diabetes-associated human leukocyte antigen (HLA) alleles are useful. Their presence predicts the development of insulin dependence in one to two years. A ratio of fasting serum C-peptide level (nmol/L) to glucose level (mmol/L) more than 11 has been suggested as a reliable predictor of possible insulin discontinuation.9 It is not known whether there is an increased risk of euglycaemic ketoacidosis, a known but rare side effect of the new SGLT2 inhibitors, but these agents would not be used on first principles. Confusingly, about one-quarter of patients with ketosis-prone diabetes have beta-cell antibodies. It may overlap with LADA.
Latent autoimmune diabetes in adults
LADA describes a condition where patients are phenotypically similar to those with type 2 diabetes and do not present in the usual fulminant way as those with unequivocal type 1 diabetes (e.g. with ketoacidosis and weight loss), but have markers of beta-cell autoimmunity (e.g. antibodies to glutamic acid decarboxylase, insulinoma-associated autoantigen 2 and zinc transporter). This is a confusing subtype and the clinical utility of diagnosing LADA has been questioned. Practically, these patients may be considered to have slowly progressive type 1 diabetes or an intermediate type between type 1 and type 2 diabetes with genetic features of both. This phenotype has also been termed ‘double diabetes’, ‘type 1.5 diabetes’ and ‘antibody-positive type 2 diabetes’.
The diagnostic criteria that have been suggested for LADA (mainly to facilitate research) are:
- presentation at age over 30 years
- presence of at least one islet cell antibody
- insulin independence for more than six months.
Initial management is with oral hypoglycaemic agents. It is difficult to predict the time to insulin requirement. Patients should be followed expectantly as progression is faster than for those with antibody-negative type 2 diabetes, although they are unlikely to decompensate quickly. Measurement of the C-peptide level may give some index of insulin secretory capacity.
A case could be made for the earlier introduction of insulin for patients with LADA, for the suggested benefit of prolonging beta-cell reserve, but this would need to be assessed on an individual basis. Similarly, it has been suggested that minimising the use of sulfonylurea drugs with their potential detrimental effect on beta-cell reserve should be considered. Patients with LADA have an increased risk of euglycaemic ketoacidosis with the use of SGLT2 inhibitors, particularly with reducing or no insulin use, and theoretically should best avoid these agents.
Mitochondrial diabetes
Mitochondrial diseases arise from mutations of either maternally inherited mitochondrial DNA (mtDNA) or less commonly, mitochondrial genes encoded by the nuclear DNA. The resultant respiratory chain defects lead to multisystem disorders that manifest in energy-dependent organs such as the heart, brain and skeletal muscle. Diabetes may also be present as mitochondria play an essential role in the release of insulin from the pancreatic beta cell via the generation of ATP, as well as a role in muscle bioenergetics affecting insulin sensitivity.
The onset of mitochondrial diabetes is often insidious, presenting commonly in the third decade. The severity and multisystem nature of mitochondrial disease may not be immediately apparent, and patients are often misdiagnosed with either type 2 or type 1 diabetes. The most common mitochondrial mutation causing diabetes is a single mtDNA point mutation (tRNALeu [m.3243A>G]). It has been estimated that 1% of all cases of diabetes are due to mutations in mitochondrial DNA.10
Clinical clues to the presence of mitochondrial diabetes are:
- maternal pattern of inheritance
- low BMI
- presence of sensorineural hearing loss, which often predates the discovery of diabetes but is often not recognised as relevant.
Myopathy, cardiac conduction abnormalities and hypertrophic cardiomyopathy may co-occur. The retinal changes of hyperpigmentation or retinal pigment atrophy may be present, but retinopathy is unusual. In contrast, there appears to be an increased vulnerability to renal and peripheral neural damage. Rarely, a serious manifestation of stroke-like episodes may occur, which present less like thromboembolic events and more like encephalopathy, with headache, visual symptoms and seizures.
Suspected mitochondrial diabetes is investigated ideally with urine or muscle biopsy samples as heteroplasmy levels (a measure of the burden of abnormal mtDNA) may be low in blood, leading to false-negative results. Laboratories in Australia will screen for common mtDNA mutations, and referral to a specialist unit is appropriate if the diagnosis is being considered.
Management of mitochondrial diabetes is similar to usual management of type 2 diabetes, but metformin should theoretically be avoided because of the increased risk of lactic acidosis. Thiazolidinediones are less desirable given their cardiac effects. Insulin is usually required early, within the first four years of the disease. Newer agents such as the dipeptidyl peptidase 4 (DPP-4) inhibitors and glucagon-like peptide-1 receptor (GLP-1) agonists could be considered. Statins may theoretically exacerbate myopathy, but a therapeutic trial should at least be considered because of their vascular and mortality benefits.
Distinguishing the different types of diabetes
A suggested diagnostic algorithm to distinguish the different types of diabetes is shown in the Flowchart. Useful resources and patient information about genetic types of diabetes are available at the Diabetes Genes website (www.diabetesgenes.org).
{kind=link}
Conclusion
In this era of personalised medicine and with an ever-widening spectrum of therapeutic agents for diabetes, the challenge for clinicians is to recognise and diagnose the various subtypes of atypical diabetes among the many patients who present with hyperglycaemia. Although atypical diabetes is rare, the diagnosis can have a significant impact on therapy and prognosis for the patient and their family. Until the advent of universal molecular testing or better biochemical subtyping of diabetes, clinicians require a high index of suspicion and a knowledge of the likely clinical phenotypes of atypical diabetes. ET
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.