Adrenal incidentaloma: key issues in screening and diagnosis
Adrenal gland disorders
Endocrine diseases
All adrenal incidentalomas should be assessed for their functional status and risk of malignancy. Newly diagnosed patients should undergo screening with a focused history, examination and biochemical tests looking for signs of excess hormone secretion. Treatment of functional adrenal incidentalomas usually involves adrenalectomy, which may improve morbidity and mortality.
- All adrenal incidentalomas should be screened clinically (history and examination) and biochemically for evidence of autonomous hormone secretion, regardless of symptoms and/or signs.
- The risk of malignancy (primary or metastatic) should be determined at diagnosis, as adrenocortical cancer is an aggressive disease. Early identification and prompt complete surgical resection offer the greatest chance of cure.
- Co-secretion of adrenal androgens and glucocorticoids is due to a primary adrenocortical cancer until proven otherwise.
- For nonfunctional, benign adenomas routine surveillance imaging and biochemistry is not needed unless new symptoms and/or signs develop concerning for hormone hypersecretion and/or malignancy.
- Patients with subclinical Cushing’s syndrome from an adrenal adenoma should be assessed individually to determine benefits of operative intervention.
An adrenal incidentaloma is defined as an adrenal mass discovered while a patient is being investigated for symptoms unrelated to an adrenal problem,1 and excluding known or suspected malignancy. Although these adrenal masses are not uncommon and usually benign, two key questions need to be addressed.
- Is it functional?
- Is it malignant?
Previous recommendations to routinely undergo several repeat CT scans have been superseded with newer guidelines based on the initial biochemical and radiological characteristics of the adrenal mass.1,2
Epidemiology
Epidemiological data on the prevalence of malignant and functional adrenal incidentalomas are variable due to the population studied and the definition of adrenal incidentaloma. The prevalence of adrenal incidentalomas reported in imaging studies for other indications is about 4%.3 However, ‘real world’ studies suggest that the pick-up rate is much lower, with fewer than 1% identified on retrospective review of imaging reports.4 The prevalence of adrenal incidentalomas increases with age, and approaches 10% in postmortem studies.5 Although most are due to adrenal adenomas, other masses in the adrenal gland include myelolipoma, cysts, haemorrhage, infiltrative or infective lesions and malignant lesions (primary and metastatic). Primary adrenocortical cancer is a rare aggressive tumour, with a reported incidence of one to two cases per million per year,6 and accounts for less than 5% of all adrenal incidentalomas.7,8 However, a recent Australian study from a major tertiary institution found a higher rate of adrenocortical cancer of 8% in 96 patients presenting with an adrenal incidentaloma.9
In regards to functional status, about 85% of adrenal incidentalomas are nonfunctioning, 9.2% autonomous cortisol secreting, 4.2% catecholamine secreting (phaeochromocytoma) and 1.6% aldosterone-producing adenomas.10 Similar data have been recently demonstrated from a New Zealand series.8 Although most adrenal incidentalomas are nonfunctional, it is essential to exclude excess hormone secretion because treatment (usually surgical excision) may improve morbidity and mortality.5
Is it functional?
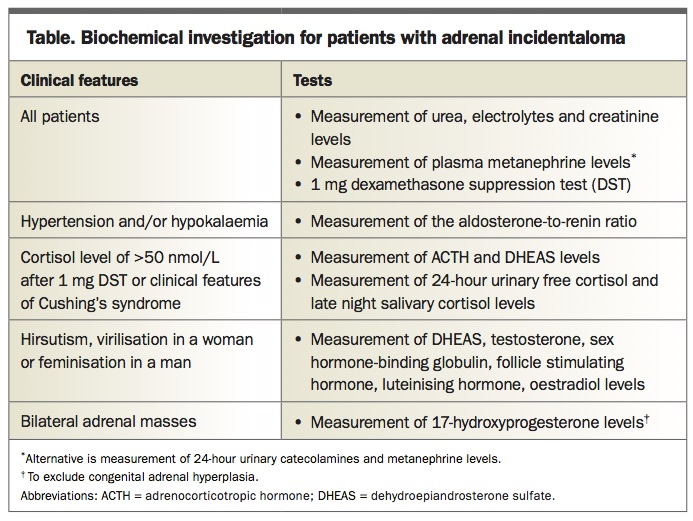
All patients with a new diagnosis of an adrenal incidentaloma should have a focused history and examination looking specifically for symptoms and/or signs of hormone excess. In the absence of any clinical abnormality, three basic screening tests should be undertaken in all patients, with additional tests depending on relevant clinical features and/or the results of the initial screening tests (Table). The three basic screening tests are measurement of urea, electrolytes and creatinine levels, measurement of plasma metanephrine levels and a 1mg dexamethasone suppression test (DST). Additionally, if hypertension and/or hypokalaemia are present, an aldosterone-to-renin ratio (ARR) should be measured.2 The main syndromes of adrenal hormone excess are briefly discussed below.
{kind=link}
Recent European guidelines suggest that if initial biochemical screening for autonomous hormone secretion from an adrenal incidentaloma is normal, then no further biochemical testing is required, unless new symptoms and/or signs develop that could be attributable to adrenal pathology.2
Phaeochromocytoma
Phaeochromocytomas are catecholamine-producing tumours that arise from the adrenal medulla and arguably are the most important functional lesion to exclude.11 Clinical signs, symptoms and imaging characteristics are variable, therefore, all patients with adrenal incidentalomas should have plasma metanephrines or alternatively 24-hour urinary fractionated catecholamines and metanephrines completed to rule out a catecholamine-producing tumour.2 Plasma metanephrines have a sensitivity of approximately 98%, which essentially excludes catecholamine excess when this test is normal.12 Certain medications including selective serotonin reuptake inhibitors, tricyclic antidepressants, monoamine oxidase inhibitors, alpha and beta blockers and some foods (e.g. caffeine) can lead to elevated plasma metanephrine levels and a false-positive test.13 To improve accuracy, plasma metanephrines should be tested after being supine for at least 20 minutes and in the fasted state.14
The most common symptoms and signs of catecholamine excess include palpitations, headache, sweating and hypertension. However, up to 50% of patients are normotensive.10 Patients with confirmed phaeochromocytoma should be managed in a specialist centre and undergo preoperative preparation, which traditionally has involved alpha with or without beta blockade. Detailed investigation and management of phaeochromocytoma has been reviewed elsewhere.15
Cortisol excess
The classic signs specific for Cushing’s syndrome are proximal myopathy, broad purple striae (>1cm in diameter), thin skin, easy bruising (evidence of current bruises), supraclavicular fat pads and unexplained osteoporosis. Other clues include unexplained deterioration in diabetes, new diabetes or hypertension.
Patients with an adrenal incidentaloma regardless of signs and/or symptoms of cortisol excess should have a 1mg DST as the initial screening test.2 A clearly normal result with an early morning (before 9.00am) post-dexamethasone cortisol level of less than 50nmol/L and no signs of Cushing’s syndrome excludes cortisol excess and no further testing is required. Patients with an adrenal incidentaloma and a 1mg DST cortisol level of more than 50nmol/L should have adrenocorticotropic hormone (ACTH), dehydroepiandrosterone sulfate (DHEAS), 24-hour urinary free cortisol and late night salivary cortisol levels measured.16 A suppressed plasma ACTH level (<2.2pmol/L or 10ng/L) or low DHEAS level (assay dependent, but about 1mcmol/L in women and 2mcmol/L in men) in this setting confirms autonomous cortisol secretion and correlates with postoperative adrenal suppression if the adenoma is resected.16,17 The urinary and salivary cortisol measures are to exclude frank cortisol excess (Cushing’s syndrome as opposed to subclinical Cushing’s syndrome). However, neither of these two tests should be used as a screening test in people with adrenal incidentalomas due to a lack of sensitivity to detect mild autonomous hypersecretion. The main exception to this would be in the case of a woman on oral oestrogen, whereby increased corticosteroid-binding globulin levels leads to elevated serum cortisol levels which may result in a false-positive DST. Medications may also affect the accuracy of the DST via induction or inhibition of the CYP3A4 enzyme, which metabolises dexamethasone.18
Subclinical Cushing’s syndrome is reported in 5 to 30% of patients with adrenal incidentalomas.19,20 Both the name and definition of subclinical Cushing’s syndrome are controversial. Failure to suppress cortisol following the 1mg DST alone is not sufficient to make the diagnosis, due to a significant false-positive rate. The recommended definition is failure to suppress cortisol following the 1mg DST to less than 50nmol/L plus evidence of autonomous secretion – either a suppressed ACTH or low DHEAS level as noted above.16,17 Subclinical Cushing’s syndrome is not a benign entity and is associated with an increased risk of cardiovascular events, infection, fractures, metabolic syndrome and mortality.19 Post-dexamethasone cortisol on suppression testing has been shown to be associated with mortality, with an increased level associated with reduced survival.21 Patients with subclinical Cushing’s syndrome should be screened for diabetes, hypertension and fractures (bone mineral density and thoracolumbar spine x-ray) to help risk stratify the patient and to determine if surgical adrenalectomy might be indicated.19
Although adrenalectomy has been shown to reduce comorbidities such as hypertension and hyperglycaemia in the setting of subclinical Cushing’s syndrome,22 no study has been adequately powered to assess if mortality is improved.16 For patients in whom surgery is not undertaken, yearly clinical assessment is recommended for up to four years, with repeat biochemistry if signs of cortisol excess develop or progress.2 Patients with subclinical or frank Cushing’s syndrome undergoing adrenalectomy must receive peri- and postoperative glucocorticoids, until the suppressed hypothalamic–pituitary–adrenal axis recovers. This may take months to years, with the duration tending to be associated with the initial severity of glucocorticoid excess.
Primary aldosteronism
The outer layer of the adrenal cortex (zona glomerulosa) produces aldosterone, which is under the control of the renin–angiotensin system, serum potassium levels and to a lesser extent ACTH.23 The main clinical manifestation of aldosterone excess is hypertension due to increased renal sodium reabsorption and subsequent water retention, with or without the presence of hypokalaemia. If hypertension and/or hypokalaemia are present in a patient with an adrenal incidentaloma, an ARR should be measured.2
False-negative ARR results can occur if renin is stimulated by a salt-restricted diet, pregnancy, malignancy, renovascular hypertension, and use of medications such as diuretics, ACE inhibitors and angiotensin receptor blockers. False-positive ARR can occur if renin is suppressed by beta blockers, alpha-methyldopa, clonidine and NSAIDs, also with renal dysfunction and advancing age.24 To accurately assess the ARR it is suggested to cease diuretics four weeks before testing and cease other interfering medications at least two weeks before testing. Verapamil slow release, hydralazine, moxonidine and prazosin can be used in the interim to control blood pressure. Patients should be advised to increase their salt intake, and hypokalaemia should be corrected before testing. To improve the sensitivity of the ARR the blood should be collected in a seated position after five to 15 minutes in the midmorning (with the patient having been ambulant for at least two hours).24 In primary aldosteronism, with chronically suppressed renin, aldosterone becomes more responsive to the circadian ACTH rhythm. Therefore, aldosterone level and subsequently the ARR ratio is higher in the morning.24
If the ARR is positive, further testing is carried out to confirm the diagnosis of autonomous aldosterone production in a specialist environment as per current Endocrine Society guidelines.25
Is it malignant?
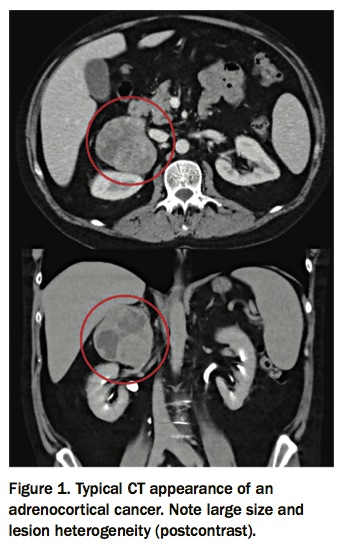
If not already performed, the initial imaging assessment is a noncontrast CT scan. Imaging characteristics of primary adrenal cancer can be nonspecific, but they tend to be associated with younger age, more than 4cm in diameter, lesion heterogeneity, more than 10 Hounsfield units (HU – the standard measure of radiodensity used in CT interpretation), postcontrast relative washout of less than 40%, necrosis or calcifications and possible evidence of vascular invasion (Figure 1).9,26 Approximately 40 to 70% of patients with adrenocortical cancer present with features of hormone excess, most commonly with hypercortisolism (50 to 80%), hyperandrogenism (40 to 60%) or a combination.6 Both elevated 24-hour urinary free cortisol and elevated serum DHEAS levels are associated with malignancy.9 Although rare, pure androgen-secreting adrenal tumours are malignant in 50% of cases, and should be identified early and resected.27 Other adrenal hormones (e.g. testosterone, androstenedione, 17-hydroxyprogesterone and oestradiol) may also be secreted and can serve as tumour markers.7,28 Any adrenal adenoma more than 4 cm in size in a patient with no history of malignancy should be considered for surgical excision. Adrenal mass biopsy is not indicated in the work-up of possible adrenocortical cancer.29 Functional imaging and/or adrenal biopsy may be helpful in patients with known malignancy to exclude adrenal metastasis. Infiltrative or metastatic lesions may be bilateral and primary malignancies known to metastasise to the adrenals include lung, breast, kidney, melanoma and lymphoma.26
{kind=link}
Benign adrenal mass
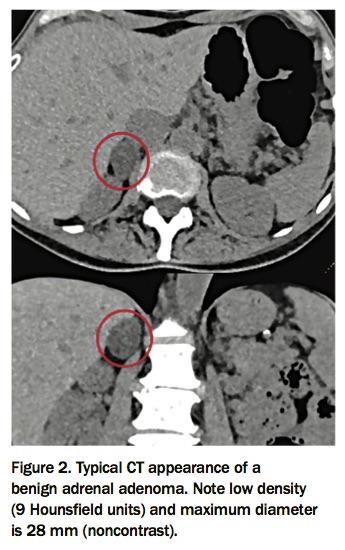
Imaging characteristics of a benign adrenal mass on noncontrast CT include a density of 10HU or less, homogenous in nature and less than 4cm in size (Figure 2).2 In a lesion with these imaging characteristics and no evidence of hormone excess, further imaging is not indicated, as these incidentalomas are rarely malignant.2
{kind=link}
Indeterminate adrenal mass
For the indeterminate adrenal mass with a density of more than 10 HU on noncontrast CT, dedicated adrenal imaging with contrast is required to assess washout characteristics to stratify malignant risk.26 Absolute washout of over 60% and relative washout of more than 40% are consistent with a lipid poor adenoma. Discussion in a multidisciplinary meeting is suggested to determine timing of repeat imaging and/or surgical intervention. Repeat adrenal imaging is recommended within six to 12 months to assess for significant adrenal mass growth (>20% increase in volume and >5mm increase in maximum diameter) and, if present, considered for surgical intervention.2 Further repeat imaging in another six to 12 months is recommended if the lesion continues to grow, but has not met the above size criteria to consider resection.2
Controversies
The recommendation from the European Society of Endocrinology that the clearly identified, less than 4cm benign nonfunctional adrenal adenoma classified at the initial diagnosis does not need follow-up imaging or biochemistry is not universally accepted.2,19 A proportion of these adrenal incidentalomas do grow, with an increase of 1cm or more in diameter reported in 3.5 to 20%. Whether or not these lesions become malignant over time is uncertain, as most lesions more than 4cm are removed. The functional status of the adrenal adenoma has also been shown to change, with new subclinical Cushing’s syndrome reported in 7.5 to 12% over follow up of more than four years.19 However, even if these adrenal masses do grow somewhat over time, the two fundamental questions as proposed initially remain.
- What is the risk of the lesion becoming malignant?
- What is the risk it will become hyperfunctioning?
The risk of malignancy developing in lesions without suspicious radiological features has been estimated to be no greater than the risk conferred by the radiation exposure from repeated CT scans.30 The second point mainly relates to the risk of new-onset subclinical Cushing’s syndrome, an entity which is being actively studied.
Summary
All adrenal incidentalomas should be assessed for their functional status and risk of malignancy at diagnosis. A suggested algorithm for work-up of a patient with an adrenal incidentaloma is shown in the Flowchart. Patients with functioning lesions such as phaeochromocytomas, adenomas causing Cushing’s syndrome and primary aldosteronism or those with nonfunctioning lesions more than 4 cm should undergo adrenalectomy. Patients with subclinical Cushing’s syndrome should be discussed in a multidisciplinary team with consideration for operative intervention if there are associated comorbidities such as hypertension or diabetes. However, definitive studies on the role of surgery in subclinical Cushing’s syndrome are required. Patients with subclinical Cushing’s syndrome should have an annual clinical examination with or without biochemical assessment for up to four years. Patients with indeterminate lesions who do not undergo surgery should have repeat adrenal imaging at six to 12 months to assess for interval growth. For the clearly nonfunctional benign adrenal adenoma less than 4cm, with a density of 10 HU or less and no other concerning features, further surveillance imaging and biochemical assessment is not necessary unless new symptoms and/or signs develop. ET