Secondary hypertension in three vignettes
Hypertension
Endocrine diseases
The immediate management and investigation of an acute endocrine presentation in general practice is discussed in this section. It is inspired by, but not based on, real patient situations.
Most patients presenting with hypertension have essential hypertension with contributing factors including age, increased body mass index, high dietary sodium and alcohol consumption. A minority develop hypertension secondary to other causes. These include medications (e.g. oral contraceptive pill, NSAIDs, nasal decongestants) and comorbidities such as renal disease, obstructive sleep apnoea and aortic coarctation. Several endocrine disorders, including primary hyperaldosteronism, phaeochromocytoma and paraganglioma (PPGL) and Cushing’s syndrome, should also be considered in the work-up of hypertension. In this review of three cases, we discuss the variable presentation of PPGL in general practice and when to investigate further.
Case study 1
A 60-year-old woman presents with a several-year history of sweating, palpitations, headache and dizziness. She had an uneventful hysterectomy under general anaesthesia 15 years ago. Two years ago, she presented to the emergency department with acute worsening of dizziness, dyspnoea at rest and right rib pain. A CT pulmonary angiogram did not reveal pulmonary embolus and she was managed with analgesia alone.
In the past year, she has experienced persistent right upper quadrant pain and 10 kg weight loss. During endoscopic retrograde cholangiopancreatography for removal of a common bile duct stone, she developed abnormal variation in blood pressure, with systolic levels above 200 mmHg and a significant postural drop of 20 mmHg while in the recovery room.
Her only medication is panadeine forte. She has no significant family history and consumes no alcohol. She sees her general practitioner for further investigation after the procedure.
What are the differential diagnoses, and what investigations would you perform?
Answer: This patient has chronic symptoms and highly suspicious labile blood pressure during anaesthesia. She should be investigated for secondary causes of hypertension. The differential diagnoses include phaeochromocytoma, renal hypertension, hyperaldosteronism, Cushing’s syndrome and other neuroendocrine tumours such as carcinoids.
Screening investigations show that the patient has elevated levels of plasma metanephrine (around five times the upper limit of normal) and normetanephrine (just above reference range), and 24-hour urinary adrenaline and metanephrine (Table). The plasma cortisol falls to appropriate levels after a 1 mg dexamethasone suppression test, making Cushing’s syndrome an unlikely diagnosis. The aldosterone to renin ratio is not elevated. The biochemical results are consistent with an adrenaline-secreting phaeochromocytoma.
{kind=link}
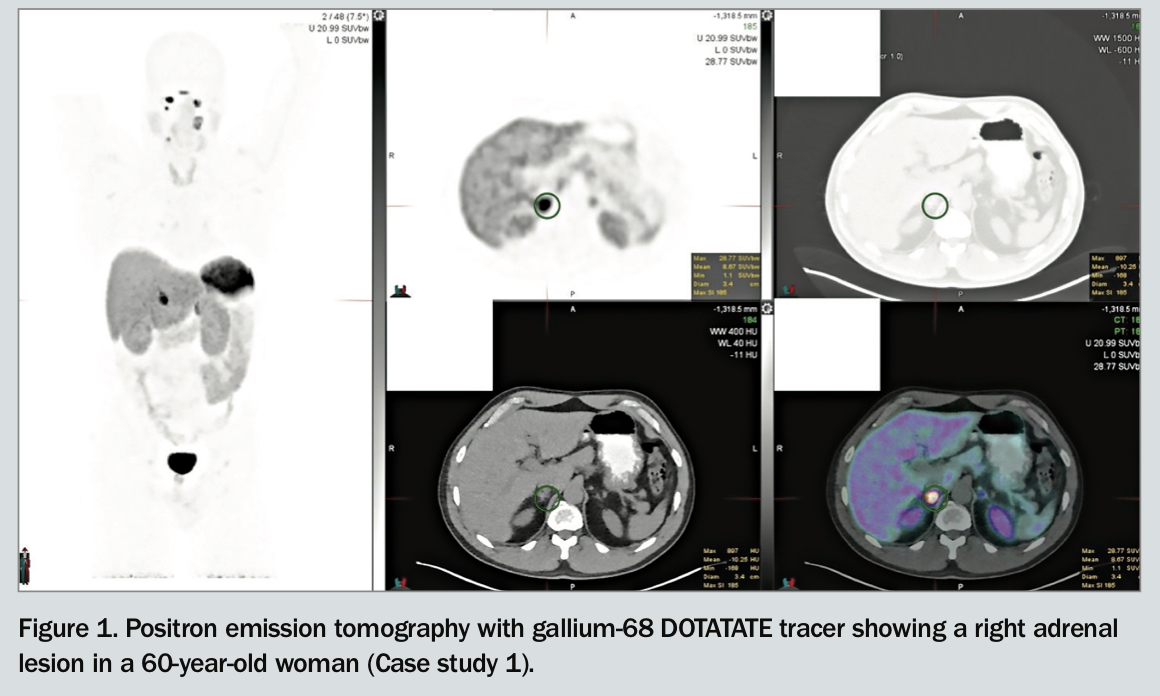
The patient undergoes abdominal CT, which shows a single right-sided 22 mm adrenal mass. A gallium-68 DOTATATE positron emission tomography scan is also performed, confirming the single right-sided avid adrenal lesion without any metastatic disease (Figure 1).
{kind=link}
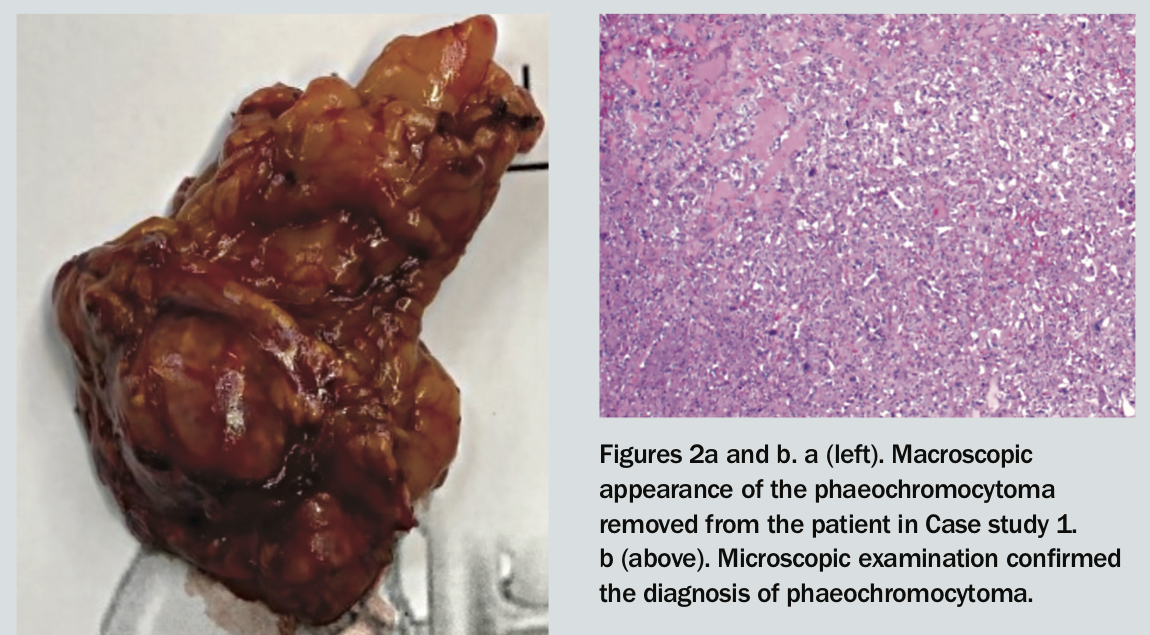
The patient undergoes laparoscopic right adrenalectomy via a retroperitoneal approach (Figure 2a). Histopathological examination confirms the diagnosis of phaeochromocytoma (Figure 2b). Her postoperative course is uncomplicated.
{kind=link}
Case study 2
A 38-year-old woman presents to the emergency department with sudden onset of severe dyspnoea and sweating while at rest. Her blood pressure is 239/172 mmHg and pulse 140 bpm and regular. Her oxygen saturation is 79% on high-flow oxygen. Clinical and x-ray findings are consistent with acute pulmonary oedema. Her blood glucose level is 21 mmol/L in the absence of any previous diabetes history. She denies any cardiac risk factors. Her past medical history is significant only for gestational hypertension three years prior, managed successfully with labetalol during pregnancy. She reports a 30 kg weight loss in preceding months, with polyuria and polydipsia.
An echocardiogram shows only mild ventricular dysfunction and left ventricular hypertrophy. Her blood pressure settles within 48 hours and she is discharged on a calcium channel blocker. Biochemical testing for catecholamine excess is performed in hospital with results pending at discharge. She presents to her general practitioner two weeks later with a copy of the discharge summary. Biochemical results are obtained from the laboratory and are consistent with catecholamine excess. Her blood pressure remains well controlled on a single agent.
What are the next steps in management and referral?
Answer: The patient’s presentation is consistent with a spontaneous adrenergic crisis. Biochemical screening shows that the patient has markedly raised plasma normetanephrine and metanephrine levels (Table). A CT scan showed a 5 cm right adrenal lesion, consistent with a phaeochromocytoma.
The history of weight loss and new diabetes fits with the biochemical diagnosis of phaeochromocytoma, despite the lack of chronic symptoms and current ease of blood pressure control. The patient should be referred to an endocrinologist. Switching to an alpha-blocker could be considered while awaiting this review.
Case study 3
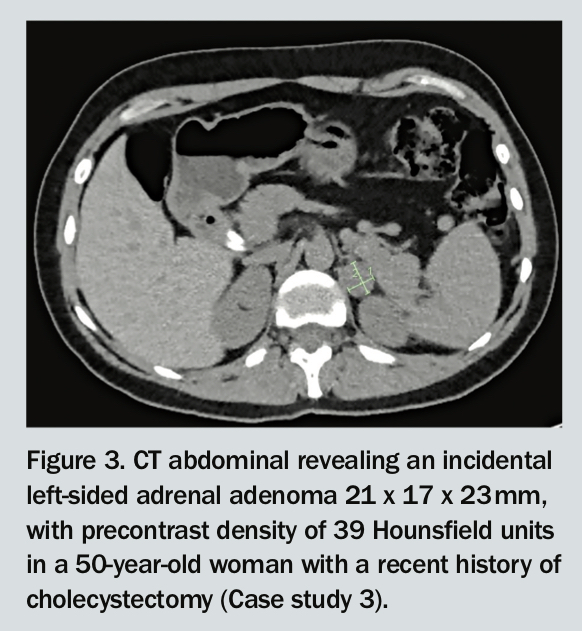
A 50-year-old woman presents with an incidental finding of a 2 cm left adrenal lesion on an abdominal CT scan, performed because of persistent epigastric discomfort (Figure 3). The lesion has a precontrast density of 39 Hounsfield units. Her past history includes a recent cholecystectomy, and bilateral oophorectomy and hysterectomy for endometriosis a year ago.
{kind=link}
On further questioning, she reports having palpitations many years prior. A 24-hour Holter monitor study has normal results. She denies any previous hypertension and had uneventful general anaesthesia during recent surgery. She has no other significant past or family history. Her current blood pressure and pulse are normal. She comes to her general practitioner for further follow up of the adrenal ‘incidentaloma’.
What are the differential diagnoses for an adrenal incidentaloma, and what investigations should be performed?
Answer: In this case, the patient presents with an adrenal incidentaloma. The main clinical considerations include determining whether the lesion is benign or malignant, and functional (hormone-secreting) or nonfunctional. A dedicated adrenal CT protocol with calculation of washout ratios can help differentiate benign adenomas from more concerning lesions. Biochemical testing can identify autonomous tumour production of catecholamines (noradrenaline, adrenaline and dopamine), aldosterone, cortisol and androgens (Table).
In this case, the patient has elevated 24-hour urinary metanephrine and adrenaline levels, around twice the upper limit of normal. Other adrenal hormone screening test results are normal. This is consistent with the left-sided adrenal phaeochromocytoma. She undergoes laparoscopic left adrenalectomy via a retroperitoneal approach and has an uncomplicated recovery.
Discussion
How do phaeochromocytoma and paraganglioma commonly present?
Phaeochromocytomas are catecholamine- secreting tumours of the chromaffin cells of the adrenal medulla. Equivalent tumours of the sympathetic ganglia are called paragangliomas. Phaeochromocytomas and paragangliomas (PPGL) may present similarly but they have distinct clinical associations and implications. Both may occur in the context of a genetic syndrome. Although predominantly benign lesions, about 10% of catecholamine-secreting tumours have metastatic potential. In addition, around 30 to 40% of these could harbour germline or somatic genetic mutations predisposing patients to PPGL. This will be covered further in sections below.
The patient in Case 1 presents with the classic triad of headache, sweating and palpitations, symptoms that occur in 51.9%, 48.85% and 58.1%, respectively, of patients with PPGL.1 Other presenting symptoms include anxiety, tremors, pallor, abdominal symptoms (nausea, vomiting and constipation), weight loss, new glucose intolerance and hypertension.2,3 Blood pressure can be variable,and may include sustained, labile or paroxysmal hypertension. Some patients present with hypotension or postural hypotension. Severe catecholamine surges can occur spontaneously or under provocation by drugs (such as general anaesthetic, beta-blockers, tricyclic antidepressants and corticosteroids), mechanical palpation during surgery or with consumption of tyramine-rich foods.4,5 Rarely, patients present with fulminant cardiomyopathy and flash pulmonary oedema, as seen in Case 2.
Case 3 represents an increasing mode of presentation – an incidental finding on imaging. Between 4 and 10% of abdominal imaging scans show an adrenal incidentaloma, with 4 to 6% of these being a phaeochromocytoma.6-8 Both MRI and CT are sensitive for diagnosis of phaeochromocytoma. Suspicious CT features include tumour size 3 cm or greater, noncontrast density of 10 Hounsfield units or more, or tumour heterogeneity.9 MRI features include isointensity with respect to the liver on T1-weighted images and hyperintensity on T2-weighted images.10
How should PPGL be investigated?
Screening
Screening tests for PPGL are appropriate in any individual with the classic triad of symptoms or sudden episodes suggestive of catecholamine excess. Additionally, people with new onset hypertension at a young age, resistant hypertension or hypertension and new-onset diabetes in the absence of other typical risk factors for diabetes should be screened. Finally, those with known phaeochromocytoma-associated familial syndromes (Hippel-Lindau syndrome, neurofibromatosis type 1, multiple endocrine neoplasia type 1), a family history of PPGL, adrenal incidentaloma with precontrast Hounsfield density of 10 units or higher, a severe hypertensive response to anaesthesia or surgery or idiopathic dilated cardiomyopathy warrant further investigation.
Biochemical tests
Initial biochemical tests include measurement of plasma metanephrine, normetanephrine and 3-methoxytyramine levels. Measurement of 24-hour urinary adrenaline, noradrenaline, dopamine and metabolite levels is an alternative. Measurement of plasma metanephrine and normetanephrine levels has high sensitivity and specificity for the diagnosis of phaeochromocytoma if collected appropriately, and is a common first-line test. It is also preferred when urine collection is unreliable, but should ideally be performed when the patient is fasting, in the supine position and after 30 minutes of rest, because of the high false-positive rate (i.e. low specificity). In nonfasting, ambulant patients, the diagnostic cut-off levels for plasma metanephrines are set higher. In the rare case of a dopamine-secreting tumour, urine and plasma catecholamine and metanephrine levels may be normal, but the urinary or plasma 3-methoxytyramine level will be raised.
In most cases, a negative result can confidently exclude a catecholamine-secreting tumour. The exception may be in patients who experience ‘spells’ due to episodic catecholamine excess. To exclude this diagnosis, the 24-hour urine collection should ideally be performed at the time of the symptomatic episode. It is also possible for small tumours detected incidentally to be secreting lower levels of catecholamines than the diagnostic cut-off. In some genetic syndromes, and particularly with paraganglioma rather than phaeochromocytoma, lesions may be nonsecretory and only detected radiologically.
False-positive results are more common, especially in the case of plasma fractionated metanephrines. Acute stress, discomfort, caffeine consumption, nicotine and benzodiazepine use or alcohol withdrawal can acutely increase catecholamine secretion. A number of medications can also affect biochemical results, particularly sympathomimetic agents, tricyclic antidepressants, antipsychotics, monoamine oxidase inhibitors and adrenergic receptor blockers. These drugs should ideally be withheld for two weeks before testing, or at least taken into account if it is not possible to stop their use.
Assuming cessation of interfering agents, results that are more than 3.5 to four times the upper limits of the reference range are usually confirmatory. Mildly raised levels are not uncommon and warrant retesting under optimised conditions, or further dynamic testing, for example, with a clonidine suppression test under the supervision of an endocrinologist.
Imaging
The next step is localisation of the tumour via imaging. Almost all (95%) of catecholamine-secreting tumours are located in the abdomen and 85% occur in the adrenal glands. Other common sites include the abdominal para-aortic areas, bladder, chest, neck and base of skull. Abdominal CT or MRI is the first-line investigation for diagnosing sporadic PPGL, as symptomatic tumours are generally large and clearly visible. CT is less expensive and has better spatial resolution, whereas MRI avoids radiation or contrast exposure. Both can distinguish phaeochromocytoma from other adrenal lesions in most cases. Modern intravenous contrast agents do not trigger hypertensive crises as the older agents did and are thus safe to use.
Functional imaging can be helpful to detect multiple tumours or metastatic disease, especially in patients with known or suspected genetic syndromes. Sporadic cases with a solitary adrenal lesion on CT or MRI do not require functional imaging before surgery. Traditionally, iodine-123-metaiodobenzylguanidine (MIBG) scintigraphy has been used to evaluate disease and plan management approaches in people who have known metastases or are at high risk of metastases due to large primary tumour size, or extra-adrenal, multifocal or recurrent disease. More recently, gallium-68 DOTATATE positron emission tomography, which detects somatostatin receptor-positive lesions, has been preferred to MIBG imaging in some centres because of its high sensitivity and specificity in detecting metastatic disease (Figure 1).
How should PPGL be managed?
Initial management
People with suspected phaeochromocytoma should avoid situations that might precipitate an adrenergic crisis. These include significant physical or emotional stress and use of drugs such as corticosteroids, metoclopramide or sympathomimetics. Importantly, beta-blockers should not be commenced before adequate alpha-blockade, as beta-2-adrenoceptors mediate vasodilatory responses. Beta-blockade will result in unopposed alpha-receptor mediated vasoconstriction, which may precipitate a hypertensive crisis.
After diagnostic confirmation, alpha-blockers are the typical first-line treatment (discussed below); however, their side-effect profile may limit long-term use. In patients awaiting surgical management who tolerate alpha-blockade poorly, calcium channel blockers are a reasonable alternative for achieving blood pressure control. Alpha-blockade will need to be initiated and uptitrated at least seven to 14 days before surgery.
Surgery after medical preparation is the prime mode of management for phaeochromocytoma. Suspected cases should be promptly referred to an endocrinologist who will involve an experienced endocrine surgeon to perform resection when the patient is adequately prepared.
Perioperative management
The main goal of medical preparation before resection of a catecholamine-secreting tumour is to prevent perioperative catecholamine secretion, which can be triggered by induction of anaesthesia and surgical manipulation. The principles are to stabilise heart rate and blood pressure fluctuations, and increase plasma volume before surgery. Firstly, patients should have adequate alpha- blockade. There are no randomised controlled trials comparing selective alpha-1 blockers such as prazosin, doxazosin or terazosin, or nonselective alpha-blockers such as phenoxybenzamine. Alpha-blockade should be started seven to 14 days before surgery, with a target blood pressure of below 130/80 mmHg when the patient is lying down and below 100 mmHg systolic when they are standing. Usual starting doses for phenoxybenzamine are 10 mg once or twice a day, increasing every two to three days to a maximum tolerated dose, or when blood pressure has been satisfactorily suppressed at 1 mg/kg. Initial doses for prazosin are 0.5 to 1 mg two to three times a day. Major side effects of alpha-blockade include nasal congestion, postural symptoms, gastrointestinal irritation and drowsiness. Patients should be reviewed every three to four days or should perform home blood pressure monitoring to assess the effectiveness of alpha-blockade. Phenoxybenzamine has a restricted PBS indication for management of phaeochromocytoma.
Once patients have adequate alpha-blockade, they should commence beta-blockade. Options include atenolol 12.5 to 25 mg two to three times a day or metoprolol 25 to 50 mg two to three times a day. The aim is to reduce the heart rate to below 80 bpm. If alpha- or beta-blockers are not tolerated, calcium channel blockers may be used (amlodipine 10 to 20 mg, nifedipine 30 to 90 mg or verapamil 180 to 450 mg daily).11,12
On the day before surgery, some patients may need to be admitted for intravenous fluid administration to expand plasma volume and avoid postoperative hypotension. Patients should maintain a high salt diet for the same purpose.
The surgical approach depends on the size of the tumour, the expertise of the surgical unit and patient factors. The three main options include minimally invasive abdominal laparoscopic adrenalectomy, open abdominal resection for tumours larger than 6 cm or a laparoscopic posterior retroperitoneal approach. The last has been associated with shorter hospital stays and reduced perioperative pain.13 After surgery, patients should be monitored for haemodynamic instability in the intensive care unit.
All three patients presented here had laparoscopic adrenalectomy and an unremarkable postoperative course. Two patients had surgery via the retroperitoneal approach. Phaeochromocytoma was confirmed via histopathology (Figure 2b). The patient in Case 2 was able to cease antihypertensive medication and had remission of her diabetes after surgery.
How should PPGL be followed up long term?
Around 17% of all cases of PPGL may recur and thus lifelong surveillance should be considered.14 This may be performed with annual biochemical tests and imaging as required.
Over 40% of PPGL tumours carry a germline or somatic mutation.15 These are divided into two main clusters.16 The first cluster involves the hypoxia-related genes, including Von Hippel Lindau disease and the paraganglioma syndromes 1 to 4 (due to mutations in the succinate dehydrogenase subunits A to D and associated enzymes including fumarate hydratase). The second cluster involves protein kinase pathways, including in RET (rearranged in transcription and accounting for multiple endocrine neoplasia type 2 syndrome), TMEM127 (controls cell growth and survival) and MAX genes, as well neurofibromatosis pathways.17 Each germline mutation is associated with its own pattern of PPGL formation, hormonal secretion, location, malignant potential and associated conditions. Genetic screening should be offered to all patients with phaeochromocytoma, and if a mutation is discovered, family tracing via genetic counselling should be undertaken. Patients and family members with germline mutations should be referred to specialised centres with experience in hereditary endocrine tumours, as screening for associated tumours is indicated. These are summarised in the evIQ guidelines.18 All three patients presented here were referred for genetic screening.
Conclusion
Hypertension is a common presenting symptom in general practice. It is important to recognise patients with secondary hypertension with reversible causes, including PPGL. The cases described highlight the variable presentations of this syndrome. GPs are well placed to perform screening investigations for suspected PPGL. Subsequent management should occur under specialist care. Practice points, including investigations for PPGL and management principles, are summarised in the Box. ET
{kind=link}
COMPETING INTERESTS: None.
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.