Investigating autoimmune polyendocrine syndromes

Autoimmune diseases
Endocrine diseases
Case scenarios are used in this section to educate doctors on the best approach to the diagnosis and management of patients with different endocrine problems. The appropriate selection of tests and correct interpretation of test results are discussed.
Autoimmune polyendocrine syndromes (APS) are a group of immune endocrinopathies, characterised by functional impairment of multiple endocrine glands due to loss of immune tolerance.1 They can also result in various nonendocrine immune disorders such as alopecia, coeliac disease, vitiligo, autoimmune gastritis and pernicious anaemia.1 They are characterised by circulating antibodies and lymphocytic infiltration of the affected organs, which eventually lead to organ failure.1 Risk of developing the component autoimmune diseases is influenced by genetic susceptibility and environmental factors. Discrete genes may be involved including the autoimmune regulator gene, AIRE, and fork-head box P3 gene, FOXP3. There is variation in presentation and symptoms, which can make diagnosis difficult. Diagnosis requires coexistence of at least two autoimmune endocrinopathies.2
These syndromes have varied presentations and can manifest sequentially at different stages during a patient’s life. An asymptomatic latent period of months to years is characterised by the presence of circulating autoantibodies. One report suggests that more than 20 years could lapse between the diagnosis of one endocrinopathy and diagnosis of another.3 Hence, long-term follow up of these patients is crucial. Management centres around monitoring and replacement of multiple hormones, along with treatment of other nonendocrine manifestations. Treatment of multiple deficiencies may be complex. The interaction of different hormonal therapies and pharmacological agents also needs to be considered.
Types of APS
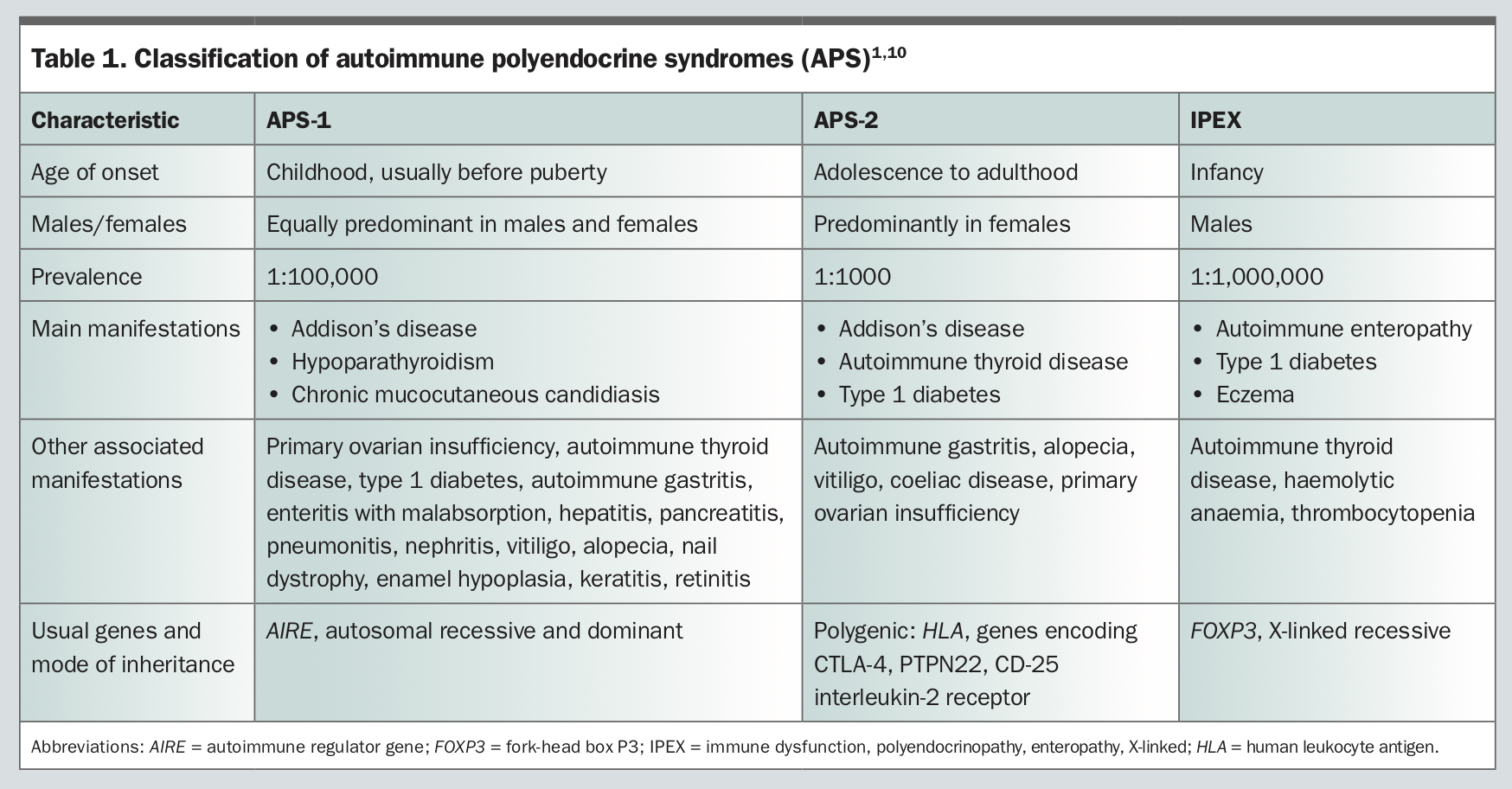
APS can be classified based on their modes of inheritance, age at presentation and characteristic patterns of disease combinations. The two major subtypes are APS-1 and APS-2. A third subtype, IPEX (immune dysfunction, polyendocrinopathy, enteropathy, X-linked), has also been described, but is rare.
APS-1
APS-1 is a rare, monogenic form, which usually presents before or during puberty. Although there are no large epidemiological studies on the prevalence of APS-1, and milder forms of APS-1 may go undiagnosed, the estimated prevalence of APS-1 is roughly 1:100,000 in most countries, with a higher prevalence in some places such as Finland (1:25,000) and Sardinia (1:14,000), and among some populations such as Iranian Jews living in Israel (1:9000) and in populations characterised by a high degree of consanguinity.1,4
APS-1 is characterised by at least two of the following three major features:1
- hypoparathyroidism
- primary adrenal insufficiency (Addison’s disease)
- chronic mucocutaneous candidiasis.
Other disease manifestations include: gonadal failure (hypergonadotrophic hypogonadism including premature ovarian failure), type 1 diabetes, autoimmune gastritis, pernicious anaemia, autoimmune hepatitis, alopecia, vitiligo, keratoconjunctivitis and asplenia (Table 1).
{kind=link}
Normally, developing T-cells with a potential reactivity for self-antigens are eliminated during early differentiation of the thymus by a process called immune tolerance. In APS-1, there is an underlying mutation of the AIRE gene, resulting in loss of immune tolerance with subsequent release of autoreactive T cells into the periphery, which then results in multiple autoimmune disorders.5 The loss of immune tolerance is thought to be in both central and peripheral pathways.6,7 More than 100 different mutations of the AIRE gene have been identified to date. APS-1 has an autosomal recessive inheritance, onset is usually before the age of 15 years and it is equally predominant in males and females.1
Recently, patients with a unique dominant negative mutation in the AIRE gene and autosomal dominant inheritance have been identified.1 These individuals have a milder form of the syndrome and can often present with pernicious anaemia, vitiligo, type 1 diabetes and autoimmune thyroid disease, which can be difficult to diagnose as they can be confused with the more common APS-2.1
APS-2
APS-2 is a more common, adult-onset, polygenic form of APS, with a higher prevalence (1:1000) than APS-1. It is characterised by at least two of the following endocrinopathies (Table 1):1
- type 1 diabetes
- autoimmune thyroid disease
- Addison’s disease.
Adrenal insufficiency is the initial manifestation in 50% of patients. The incidence is higher among females. Family members across multiple generations are often affected.3 The pathogenesis is less clear and involves both human leucocyte antigen (HLA) and non-HLA genes. Alleles of HLA determine the targeting of specific tissues by autoreactive T cells, which leads to organ-specific autoimmunity as a result of loss of tolerance. Non-HLA genes also contribute to autoimmunity and, depending on the polymorphism, potentially predispose to a loss of tolerance of the organ involved.5 There are various classifications of APS-2 in the literature based on combinations of component diseases.8,9 These different classifications can be confusing so, recently, all of these have been collectively termed as APS 2.8
IPEX
IPEX is a rare and fulminant form of APS, characterised by early-onset type 1 diabetes, autoimmune enteropathy with intractable diarrhoea, malabsorption and dermatitis (Table 1). It is caused by mutations in the FOXP3 gene, which results in the absence or dysfunction of regulatory T cells. Many features of IPEX overlap with APS-1, but the presentation of IPEX is usually in infancy, only in males and is often fatal if not promptly treated.1
Diagnosis and management
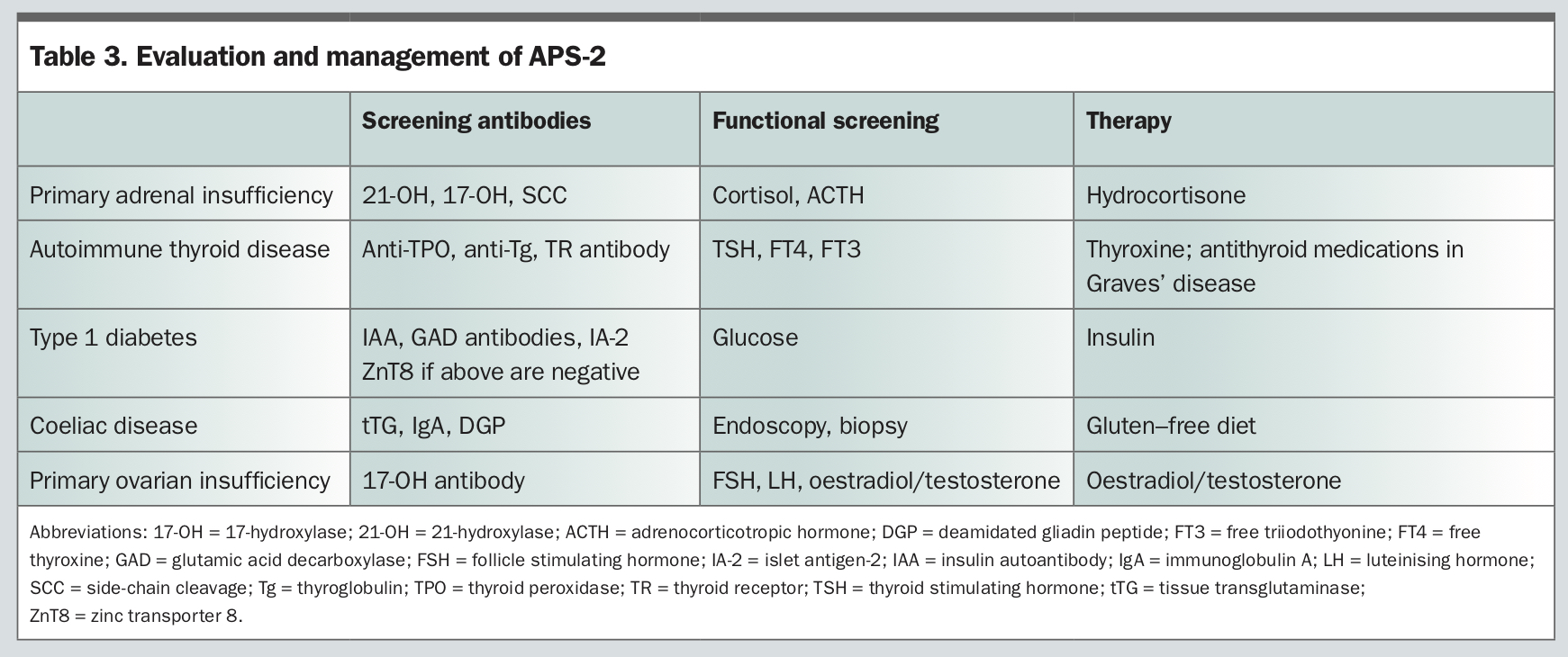
Diagnostic work-up of patients with APS includes assessment of endocrine function and consideration of serological screening for relevant organ-specific antibodies (Table 2 and Table 3). There is a clear association between the presence of organ-specific autoantibodies and progression to disease.9 Presymptomatic recognition of autoimmune disease minimises associated morbidity and mortality.9 In the presence of one endocrine manifestation, it is important to screen long term for the development of others, particularly Addison’s disease and type 1 diabetes. The clinical significance of classifying APS lies in screening patients and first-degree relatives for associated autoimmune diseases, as one in seven first-degree relatives have an unrecognised endocrine disorder.5 Patients should be counselled about the symptoms of other autoimmune disorders for which they are at high risk. Management includes:
{kind=link}
{kind=link}
- hormone replacement therapy as required
- monitoring for the development of other endocrine and nonendocrine manifestations and their treatment (Table 2 and Table 3).
Patients with APS-1 are best managed by a multidisciplinary team led by an endocrinologist or paediatric endocrinologist. It is recommended that patients have two follow-up visits a year and asymptomatic carriers be followed up at least annually.1 Children should be followed up regularly by their paediatric endocrinologist until later adulthood.
Case 1. APS-1
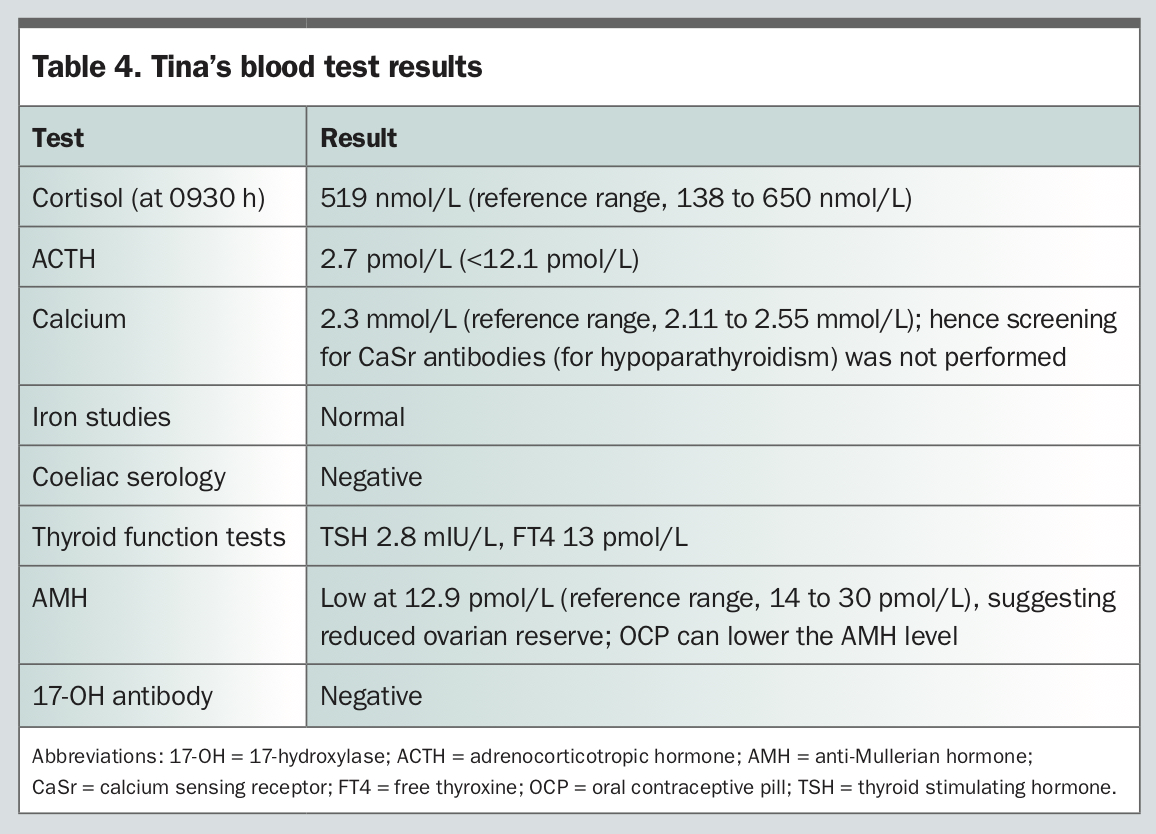
Tina is a 25-year-old woman who is concerned about her severe fatigue, which has significant impact on activities of daily living. She was diagnosed with type 1 diabetes at the age of 2 years. Over the following years, she developed multiple other autoimmune disorders: Hashimoto’s thyroiditis at age 10 years (positive anti-TPO 559 IU/L); chronic idiopathic urticaria and angioedema; and a recent diagnosis of pernicious anaemia. Her brother also has type 1 diabetes. Tina’s current medications include thyroxine 100 mcg daily, immunosuppressive therapy with mycophenolate 500 mg twice a day, a leukotriene receptor antagonist, montelukast, for chronic idiopathic urticaria and the oral contraceptive pill (OCP) for regulation of menstrual cycles.
{kind=link}
{kind=link}
Case 2. APS-2
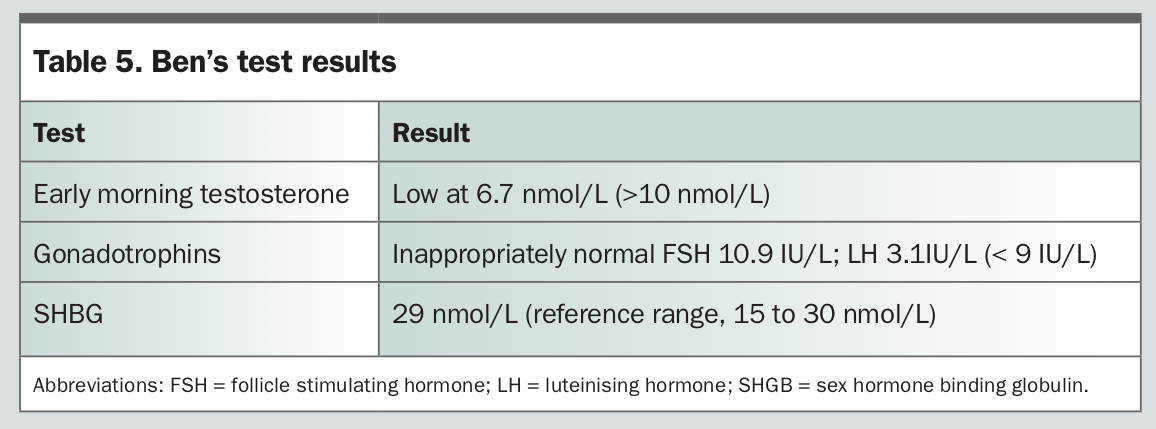
Ben is a 31-year-old man with well-controlled type 1 diabetes (glutamic acid decarboxylase [GAD] 831IU/mL, negative islet antigen 2 [IA-2] and islet cell antibody counts) and autoimmune thyroiditis. He presents for evaluation of infertility. Ben had normal pubertal development of secondary sexual characteristics and denied a history of testicular trauma or surgery. On examination, his body mass index is 38.1 kg/m2, he has an underdeveloped penis (less than 4 cm) and bilateral small testes (8 mL in volume).
{kind=link}
Subsequent investigations should include a complete pituitary panel (including prolactin) and a pituitary MRI, to rule out a secondary cause of hypogonadism, both of which had unremarkable results in Ben’s case. Other investigations that should be considered are semen analysis, which shows azoospermia in Ben’s case, and a karyotype for exclusion of chromosomal abnormalities which, in this case, was normal. A microdeletion study may be considered to look for genetic causes of impaired spermatogenesis.
Case 3. APS-2
Deborah is a 67-year-old postmenopausal woman, who is referred to our endocrinology service with a past diagnosis of Addison’s disease and hypothyroidism, and recent diagnosis of diabetes and iron deficiency. She reports symptoms of lethargy and is noted to be iron deficient. Her diabetes has been managed with oral hypoglycaemic agents alone to date.
- Screening tests:
– intrinsic factor and gastric parietal cell antibody (for pernicious anaemia)
– total immunoglobulin A (IgA), tTG (tissue transglutaminase)-IgA, deamidated gliadin peptide (DGP) antibody (for coeliac disease)
– calcium sensing receptor (CaSR) antibody (for hypoparathyroidism)
glutamic acid decarboxylase (GAD), islet antigen-2 (IA-2), zinc transporter 8 antibody (ZnT8Ab), islet cell antibody - Functional tests:
– thyroid stimulating hormone (TSH), free thyroxine (FT4), free triiodothyronine (FT3)
_ glycated haemoglobin (HbA1c), glucose
– iron studies, serum vitamin B12 and folate
– corrected calcium
Table 6 shows Deborah’s blood test results. The results indicate Deborah has late-onset autoimmune type 1 diabetes. Iron deficiency is confirmed. Positive intrinsic and gastric parietal cell antibodies suggest pernicious anaemia, although her serum vitamin B12 level is normal.
{kind=link}
Conclusion
{kind=link}
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.