Cystic fibrosis-related diabetes: early diagnosis and management

Cystic fibrosis
Diabetes medicines
Cystic fibrosis-related diabetes (CFRD) can present slowly and may significantly impair patient health and survival. Regular screening of glucose levels and insulin therapy are cornerstones of management. Patient education is important to help informed decision-making. GPs can play an important role in the early identification and management of CFRD as part of a multidisciplinary team.
- As a part of the broader multidisciplinary cystic fibrosis (CF) team, GPs should be aware of the symptoms and signs of emerging CF-related diabetes (CFRD).
- Initially these may include gradual unexplained weight and/or lung function decline, more frequent respiratory exacerbations and infections and, later, the classic symptoms of diabetes – polyuria and polydipsia. Ketoacidosis may occur but is rare.
- Insulin therapy to correct insulin deficiency is the mainstay of treatment. The high-calorie CF diet is continued, and is guided by a dietitian with expertise inCF.
- Self-monitoring of blood glucose levels using finger-prick testing is needed. Continuous glucose monitoring is also increasingly used and provides real-time interstitial glucose levels to help the patient make informed decisions on dosing, activity and diet. Appropriate education is needed.
- Current diagnostic thresholds for CFRD are taken from type 2 diabetes, and patients with CFRD often show a decline in weight and lung function before diagnosis.
- CF modulatory therapies may delay the timing and expression of insulin deficiency in patients with CF, but more research is needed in this area.
Cystic fibrosis (CF) is a common, multisystem autosomal recessive disease, affecting the CF transmembrane conductance regulator (CFTR) protein and, if untreated, causes early death due to respiratory and pancreatic disease.1 Recent decades have seen a dramatic increase in life expectancy, but this has come at the cost of a high treatment burden for patients and requires intensive support from a multidisciplinary CF team. The GP can help the CF team promote health and recognise emerging complications in affected patients.
Diagnosis
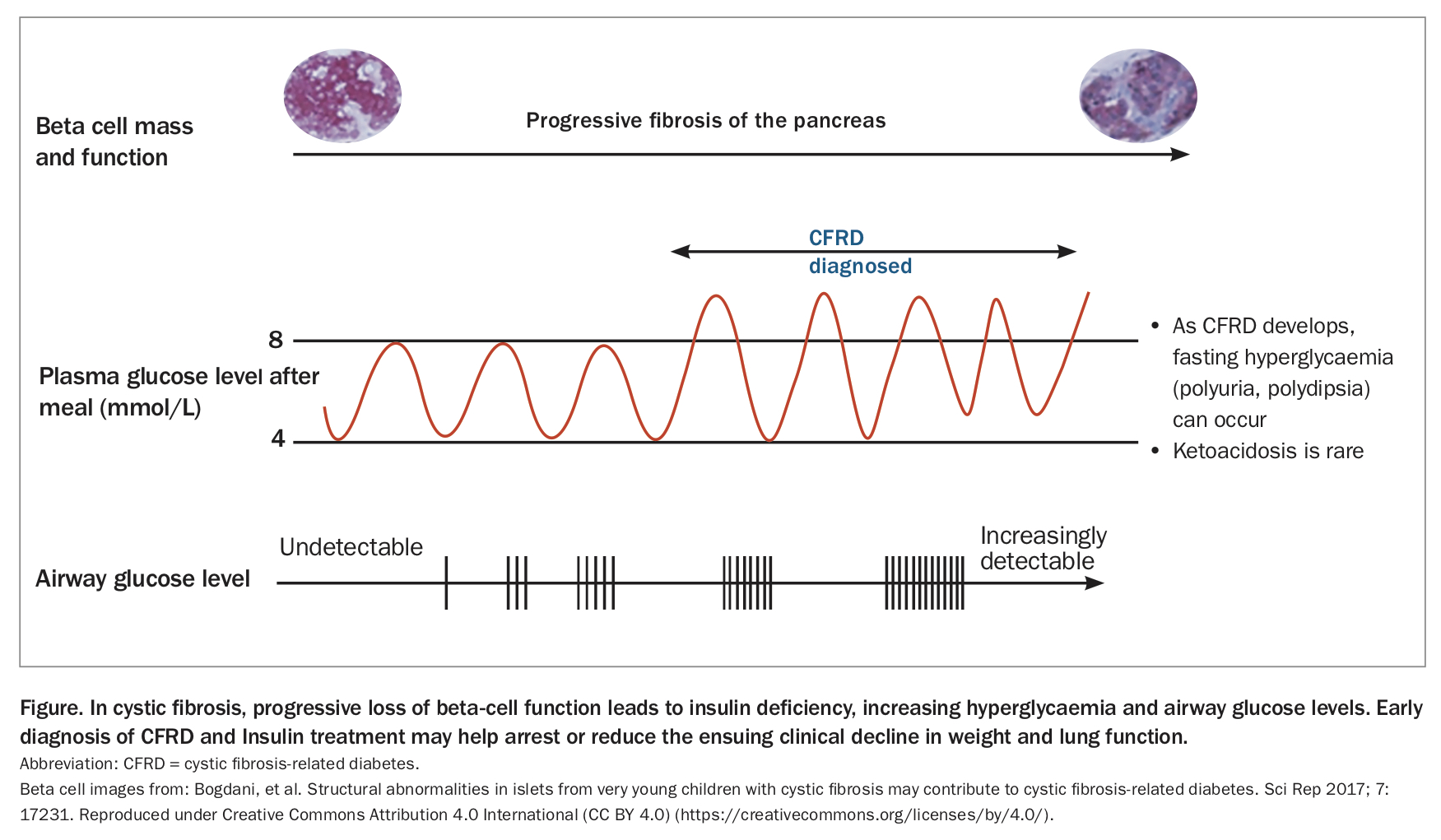
Most patients with CF are diagnosed by newborn screening and around 85% have exocrine pancreatic insufficiency at diagnosis. One of the most common complications that patients with CF will subsequently encounter is CFRD, which often presents insidiously.2 It results from the gradual reduction in insulin secreting ability, due to the underlying pancreatic disease of CF. In CF, gradual pancreatic inflammation and fibrosis often result in impaired insulin secretion, through loss of number and function of insulin-secreting beta cells. Insulin is a major anabolic hormone and its gradual loss can cause poor weight gain and, in childhood, impaired linear growth.3 Loss of anabolic function may also lead to weakened respiratory muscle function and lung function decline. Weight and lung function decline are associated with early mortality.
As insulin deficiency progresses, beta cells struggle to achieve normoglycaemia, and glucose excursions after meals become common, resulting in frequent hyperglycaemia (Figure). When blood glucose levels rise above 8 mmol/L, airway glucose levels become detectable and this can promote the growth of pathogens within the lung.4 More frequent respiratory exacerbations and infections may then occur, leading to raised inflammatory cytokine levels and release of counter-regulatory hormones such as cortisol, which in turn cause further hyperglycaemia. This results in a vicious spiral of declining clinical status and increasing hyperglycaemia. Given that in healthy individuals blood glucose levels rarely rise above 8.3 mmol/L, there is considerable interest in the nutritional and respiratory implications of these early glucose abnormalities.5 The risks and benefits of early insulin treatment (before the development of CFRD) are being investigated in the Cystic Fibrosis Insulin Deficiency Early Action (CF-IDEA) Trial (clinicaltrials.gov NCT01100892).
{kind=link}
Pancreatic beta-cell function is particularly taxed when there is increased requirement for insulin secretion. Periods of increased risk of hyperglycaemia include puberty, systemic illness and during treatment with systemic glucocorticoids.2
CFRD can develop at any age, including in infancy, but prevalence increases with age, affecting around 19% of patients during adolescence and 50% in adulthood.6 Patients may eventually present with classic features of diabetes relating to prolonged sustained hyperglycaemia, namely polyuria and polydipsia. As with type 1 diabetes mellitus, ketoacidosis may occur in patients with CFRD as a result of insulin deficiency, but is more rare because of a slower decline in beta cell mass. Other common presentations of, and recommended diagnostic tests and criteria for, CFRD are listed in the Box.2,3,7,8
{kind=link}
Given the late presentation of polyuria and polydipsia and variable clinical features of catabolic and respiratory decline in CFRD, many CF centres implement routine annual oral glucose tolerance tests for patients aged 10 years or older to diagnose CFRD. Even with routine screening, weight and lung function decline are common before the diagnosis of CFRD is made.9,10 The diagnostic criteria for CFRD are based on those for type 2 diabetes and are designed to detect microvascular complications such as retinopathy. They may be insensitive in detecting weight and lung function decline in patients with CF.3,9-11
Management
Insulin treatment to replace deficient insulin secretion is the mainstay of treatment for CFRD, with very limited data on alternative experimental therapies such as incretin-based therapies.2,12 People with CF have high caloric requirements, therefore, in the presence of CFRD, restricting dietary carbohydrates should be avoided as it runs the risk of reduced caloric intake and catabolism. This is an important difference from the treatment of type 2 diabetes, and expert nutritional guidance should be provided by a dietitian working within the multidisciplinary CF team. Once diagnosed with hyperglycaemia or CFRD, patients are started on self-monitoring of blood glucose using finger-prick testing. In Australia, blood glucose monitoring is subsidised on the National Diabetes Services Scheme (NDSS) for those on insulin treatment. More recently, continuous glucose monitoring (CGM) has been subsidised for children and young people with CF aged under 21 years. There are currently limited published data on CGM outcomes in patients with CF.13
In contrast to blood glucose testing, CGM measures glucose levels in subcutaneous interstitial fluid.14 Changes in interstitial fluid glucose levels may lag behind blood glucose level changes by several minutes.15 CGM readings can also be inaccurate, and patients should confirm results with a finger-prick test if uncertain (e.g. if they feel symptomatically hypoglycaemic, but the CGM shows a normal reading). CGM generates a lot of data on real-time glucose levels, and education for families and patients on how to interpret these and make correct management decisions is vital. Education also involves training on hypoglycaemia management, including administering intramuscular glucagon injections for emergency treatment of severe hypoglycaemia, such as if a patient is unconscious and unable to eat or drink to correct hypoglycaemia. In clinical practice, the aim is for tight glycaemic management in patients with CFRD, ideally maintaining glucose levels between 4 and 8 mmol/L at all times. To achieve these glucose levels, patients often require regular insulin adjustment with guidance by clinic staff, and comprehensive education to be able to make sound dosing decisions based on diet, activity, illness or concurrent glucocorticoid use.
Insulin therapy is usually given by subcutaneous injection and, in the early stages of dysglycaemia, a low-dose basal insulin once a day (e.g. insulin detemir or insulin glargine) may be all that is needed to normalise glucose levels (Flowchart).16 Dose escalation and addition of rapid-acting insulin before a meal may be needed if a patient’s glucose levels worsen, such as in systemic illness, during periods of glucocorticoid use, in puberty or as duration of diabetes lengthens. Some patients use continuous subcutaneous insulin infusion (insulin pump therapy), which requires intensive effort by the patient or family (and support from a multidisciplinary team). Patients are required to administer boluses of rapid-acting insulin at all meal and snack times and to correct high glucose levels during the day or night. Automated systems are used to manage type 1 diabetes mellitus, but whether the algorithms used by these systems will work as effectively in people with CFRD is unknown.
An important aspect of managing patients on insulin therapy is to ensure lipohypertrophy does not develop at injection sites because of insufficient rotation of injection locations. The GP can check on this, as well as on adherence to the insulin and glucose monitoring regimen.
CF modulator therapies alter the function of the CFTR and some, such as ivacaftor, have been shown to improve glycaemia in small case series studies. As these agents become increasingly available, effects on patients with established or emerging CFRD will need to be observed.17
Conclusion
CFRD often emerges slowly and can have a significant adverse impact on clinical status in patients with CF, including weight, lung function and respiratory infections. The clinical presentation can be insidious and delays in diagnosis and treatment can lead to a major decline in health status. The GP can play an important role in recognising the emergence of CFRD, and in addressing this clinical decline by appropriately supporting the patient’s diabetes management plan. ET
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.