Chronic musculoskeletal disorders due to FGF23-mediated hypophosphataemia

Bone diseases
Parathyroid disorders
Fibroblast growth factor 23 (FGF23) is a key hormonal regulator of renal phosphate excretion that, in excess, can lead to hypophosphataemia and chronic musculoskeletal symptoms, rickets or osteomalacia. Conditions that cause FGF23 excess can be inherited, such as X-linked hypophosphataemic rickets, or acquired, such as tumours and intravenous iron infusions. These conditions are often misdiagnosed unless serum phosphate is measured and the significance of hypophosphataemia recognised.
- Measurement of the serum phosphate level is crucial for evaluating patients with musculoskeletal complaints; failure to perform this test often delays diagnosis in patients with X-linked hypophosphataemic rickets (XLH) and tumour-induced osteomalacia.
- A thorough history and clinical examination are important; hypophosphataemia is often longstanding and undiagnosed.
- Assessment of tubular reabsorption of phosphate is key to determining whether hypophosphataemia is due to renal phosphate wasting, which should be considered if hypophosphataemia is otherwise unexplained, chronic or associated with features of osteomalacia (bone pain, proximal weakness, recurrent stress fractures and mobility or functional decline).
- Management of hypophosphataemia mediated by fibroblast growth factor 23 (FGF23) involves a multidisciplinary approach and multiple daily dosing with phosphate and calcitriol.
- Burosumab (anti-FGF23 monoclonal antibody) is an effective therapy for XLH and promising in acquired FGF23 excess disorders such as tumour-induced and iron infusion-associated osteomalacia.
Phosphate (PO4) is an essential anion required for energy metabolism, synthesis of cell membranes and nucleotides, and mineralisation of bones and teeth. Hypophosphataemia can present clinically as an acute syndrome, such as cardiomyopathy and rhabdomyolysis; or a chronic musculoskeletal disorder, such as proximal myopathy and failure of bone mineralisation, leading to rickets (if it occurs before growth plate closure) or osteomalacia (if it occurs after growth plate closure).
Hypophosphataemia is most often caused by impaired gastrointestinal absorption, often in association with malabsorption of other nutrients, or transcellular phosphate shifts, which are common in hospital patients. However, isolated hypophosphataemia is primarily due to excess renal phosphate clearance, termed renal phosphate wasting. A key hormonal regulator of renal phosphate excretion is fibroblast growth factor 23 (FGF23), which can be produced in excess in a range of inherited and acquired conditions, leading to renal phosphate wasting.
This article discusses the causes of FGF23-mediated renal phosphate wasting, their clinical features, diagnosis and management. Three case studies highlight causes that are often misdiagnosed or simply overlooked in patients with chronic debilitating musculoskeletal symptoms. Confirming hypophosphataemia and renal phosphate wasting and establishing the correct diagnosis allowed appropriate management and an overwhelming improvement in the well-being and function of these patients.
Phosphate homeostasis
The plasma phosphate concentration shows a marked diurnal rhythm; it is lowest in the morning and highest in the evening. The normal serum phosphate range is age-related:
- 0.8 to 1.5 mmol/L in adults
- 0.9 to 1.8 mmol/L at ages 1 to 16 years
- 1.3 to 2.6 mmol/L at age under 1 year.
The determinants of serum phosphate include dietary intake, gastrointestinal absorption, cellular shifts and renal excretion. Key hormonal regulators of serum phosphate include:
- 1,25-dihydroxyvitamin D3 (calcitriol), which promotes gastrointestinal phosphate absorption
- parathyroid hormone (PTH) and FGF23, which promote renal phosphate excretion.
Phosphate and the gastrointestinal tract
Phosphate is present in high concentrations in many foods, especially dairy products, meat and vegetables. Thus hypophosphataemia due to reduced dietary phosphate intake is uncommon.1
Phosphate is absorbed throughout the small intestine and colon by an active and a passive pathway. Active transport occurs via sodium-phosphate cotransporter 2b (NPT2b) in the brush border of enterocytes; its expression is increased by 1,25-dihydroxyvitamin D3. In people with vitamin D deficiency, only 50 to 60% of phosphate can be absorbed from ingested food.
Conversion of 25-hydroxyvitamin D3 to the active form 1,25-dihydroxyvitamin D3 is catalysed by the enzyme 1-alfa-hydroxylase. PTH increases expression of this enzyme, indirectly increasing gastrointestinal phosphate absorption. Conversely, FGF23 decreases expression of 1-alfa-hydroxylase and also stimulates the enzyme 24-hydroxylase, which degrades 1,25-dihydroxyvitamin D3, thereby indirectly decreasing gastrointestinal phosphate absorption.2
Phosphate and the kidneys
About 90% of the phosphate filtered by the kidneys is reabsorbed, mostly in the proximal convoluted tubule. Phosphate reabsorption is mediated by sodium-phosphate cotransporters 2a and 2c (NPT2a and NPT2c). PTH reduces expression of these cotransporters, and FGF23 promotes their degradation, thereby decreasing renal phosphate reabsorption.2
FGF23 is released primarily by osteocytes. Its expression is stimulated by a high dietary phosphate intake and high serum phosphate and 1,25-dihydroxyvitamin D3 concentrations. Its expression is inhibited by the products of phosphate regulating gene with homologies to endopeptidases on the X chromosome (PHEX) and the dentin matrix acidic phosphoprotein-1 (DMP1) gene. Hence, inactivating mutations in PHEX and DMP1 result in excess circulating FGF23. After translation, FGF23 also undergoes significant modification and cleavage; it can be protected from this cleavage by the protein GALNT3 or marked for destruction by the protein FAM20C. Hence, circulating FGF23 concentrations are determined by gene expression and also post-translational modification and cleavage (Figure 1).3-5
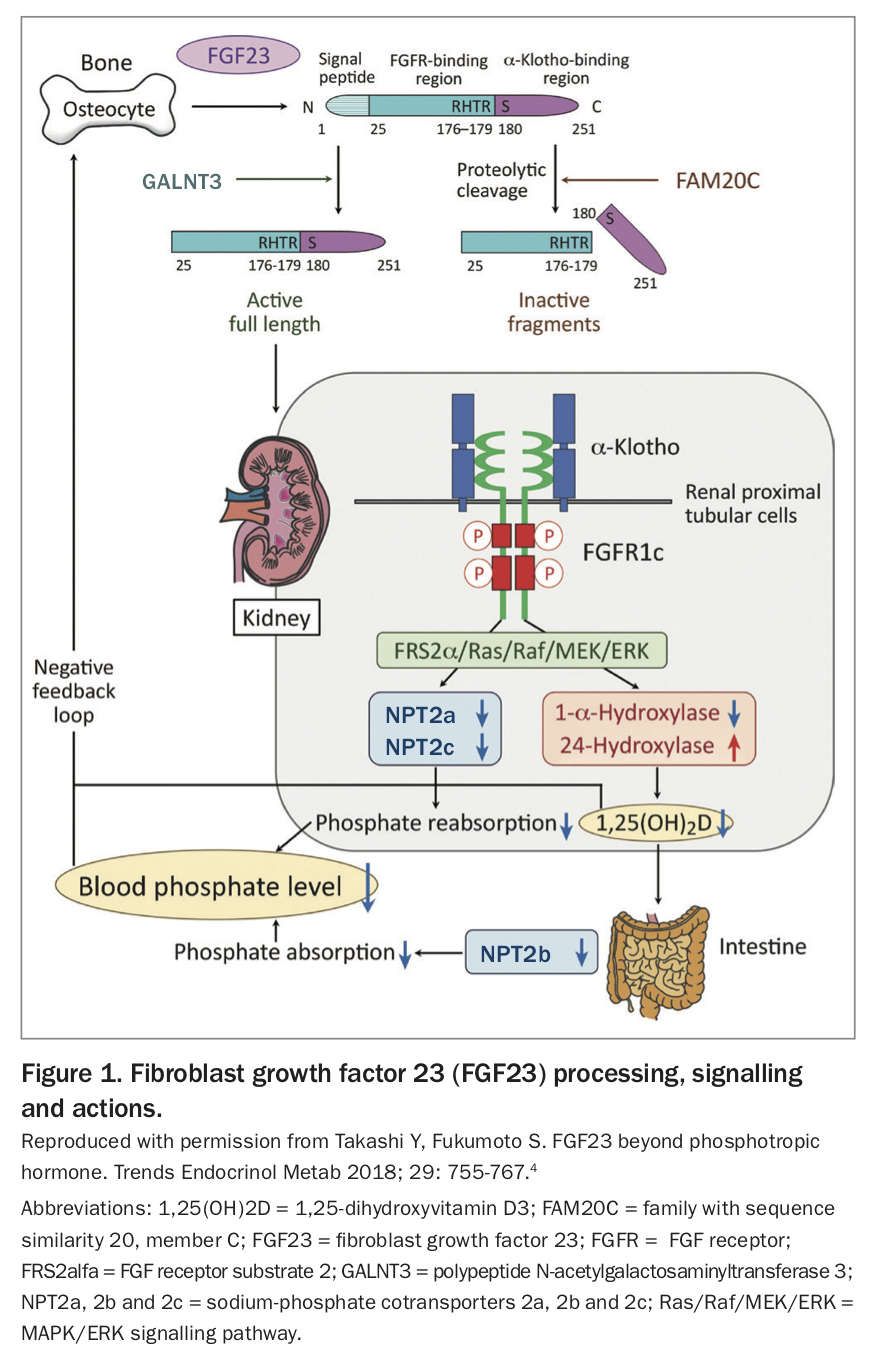{kind=link}
Diagnosis of hypophosphataemia
In adults, a plasma phosphate concentration less than 0.80 mmol/L is defined as hypophosphataemia, and less than 0.30 mmol/L as severe hypophosphataemia.
Cause of hypophosphataemia
Possible causes of hypophosphataemia are listed in Box 1. The diagnostic pathway to differentiate the various causes of hypophosphataemia is summarised in the Flowchart.
{kind=link}
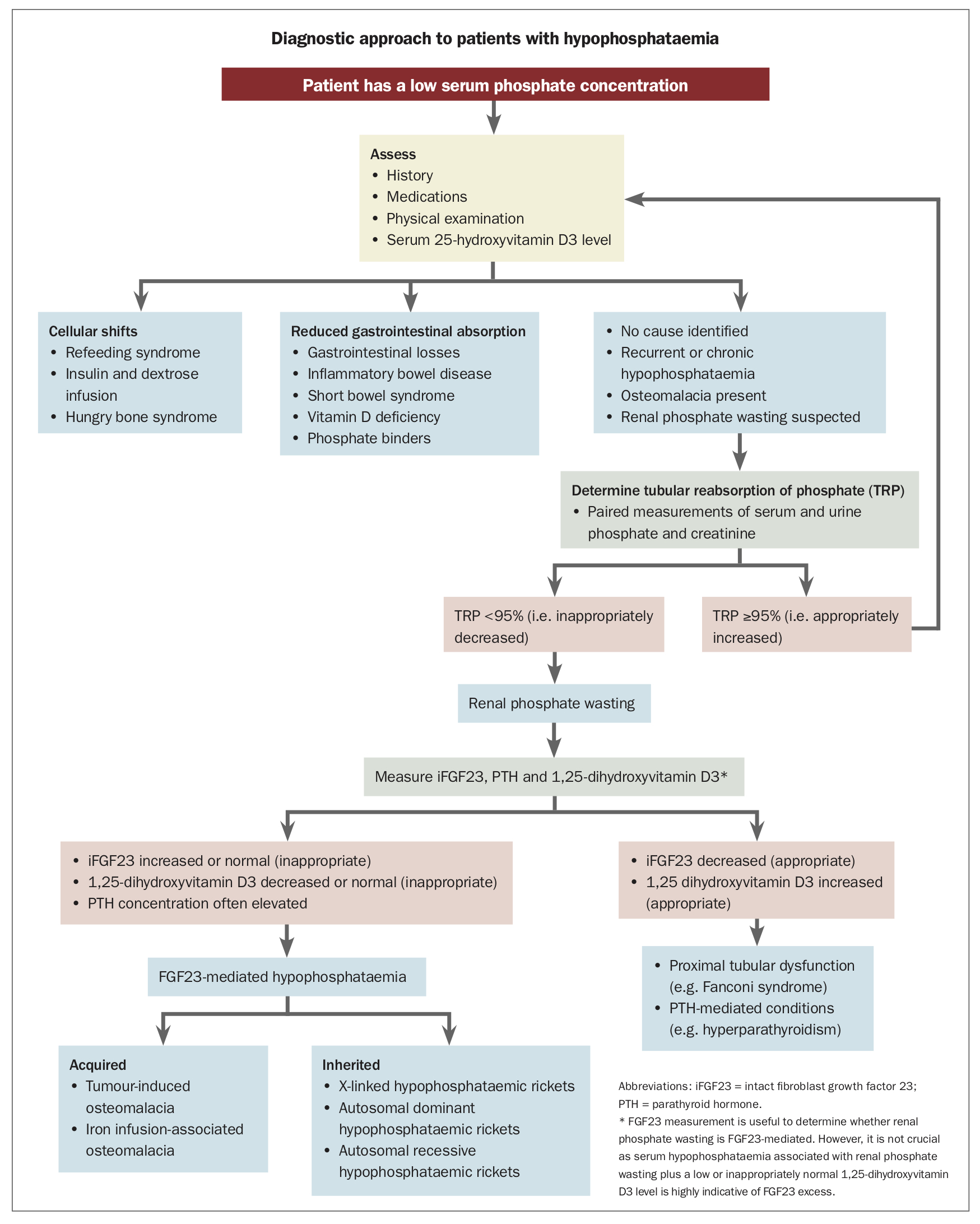{kind=link}
Most cases of hypophosphataemia are secondary to cellular shifts or impaired gastrointestinal absorption.1 Hypophosphataemia is seen in about 2 to 5% of hospital inpatients, commonly as a result of acute transcellular phosphate shifts. Abnormalities in gastrointestinal phosphate absorption are often associated with calcium and vitamin D deficiency and potentially other nutritional deficits. Detailed history taking, physical examination and measurement of the serum 25-hydroxyvitamin D3 concentration are usually sufficient to identify these causes.
If no clear cause of hypophosphataemia is identified or it is chronic or associated with clinical features of osteomalacia then renal phosphate wasting should be excluded. Determination of the tubular reabsorption of phosphate (TRP) can confirm abnormal regulation of renal phosphate excretion.
Assessment of tubular reabsorption of phosphate
TRP determination requires simultaneous measurement of fasting serum and spot urine phosphate and creatinine levels. Spot urine samples for phosphate determination should be obtained after patients have been off phosphate supplements for at least 24 hours. TRP is calculated from the equation:
TRP = 100 x [1 – (spot urine phosphate level x serum creatinine level)/(serum phosphate level x spot urine creatinine level)]
Automated calculators to determine TRP are available online (e.g. www.scymed.com/en/smnxps/pshpd274.htm). Ensure correct units are used in the calculator (incorrect units are a common error).
When the serum phosphate concentration is normal, the normal range for TRP is 85 to 95%. In cases of hypophosphataemia, renal phosphate reabsorption should be maximal, with a TRP of 95% or higher. Reduced TRP in the presence of hypophosphataemia indicates a renal defect in phosphate reabsorption (renal phosphate wasting).
Hypophosphataemic osteomalacia and rickets
Hypophosphataemia due to renal phosphate wasting is often chronic and overlooked and may result in myopathy and metabolic bone disorders, termed hypophosphataemic osteomalacia or rickets. This condition is defined by a low serum phosphate level (less than 0.80 mmol/L) plus an inappropriately reduced TRP (less than 95%). Unlike nutritional rickets, hypophosphataemic osteomalacia and rickets cannot be cured with nutritional vitamin D supplementation.
Hypophosphataemic osteomalacia or rickets may be either FGF23 or non-FGF23-mediated. FGF23-mediated renal phosphate wasting is further characterised by elevated or inappropriately normal FGF23 concentrations and suppressed 1,25-dihydroxyvitamin D3 concentrations. It occurs in a range of inherited and acquired conditions.
Inherited causes of FGF23-mediated hypophosphataemia
Several inherited syndromes can cause FGF23-mediated hypophosphataemia. Clues to an inherited cause include disproportionate short stature, early onset, positive family history and skeletal deformities such as bowing of the femur and tibia.6
X-linked hypophosphataemic rickets
The most common inherited cause of renal phosphate wasting is X-linked hypophosphataemic rickets (XLH), with an incidence of one in 20,000 live births. It is caused by an inactivating PHEX mutation that increases the expression of FGF23. Clinical characteristics include rickets, disproportionate short stature, osteomalacia and odontomalacia.
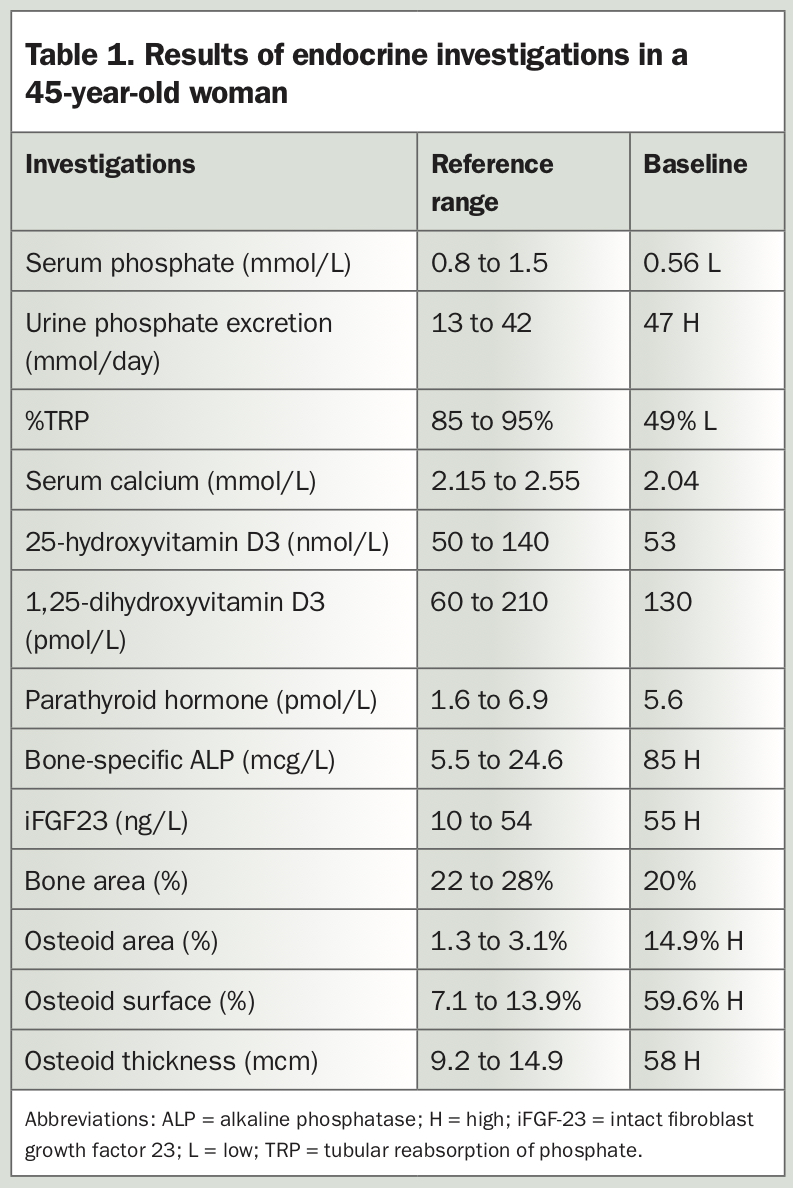
XLH usually presents in children within the first two years of life. Delayed diagnosis in adulthood is often the result of the diversity of clinical manifestations and rarity of the disease.6 The different clinical presentations in children and adults are shown in Figure 2.7 The case of a 45-year-old woman with XLH misdiagnosed as ankylosing spondylitis is described in Box 2 and Table 1.
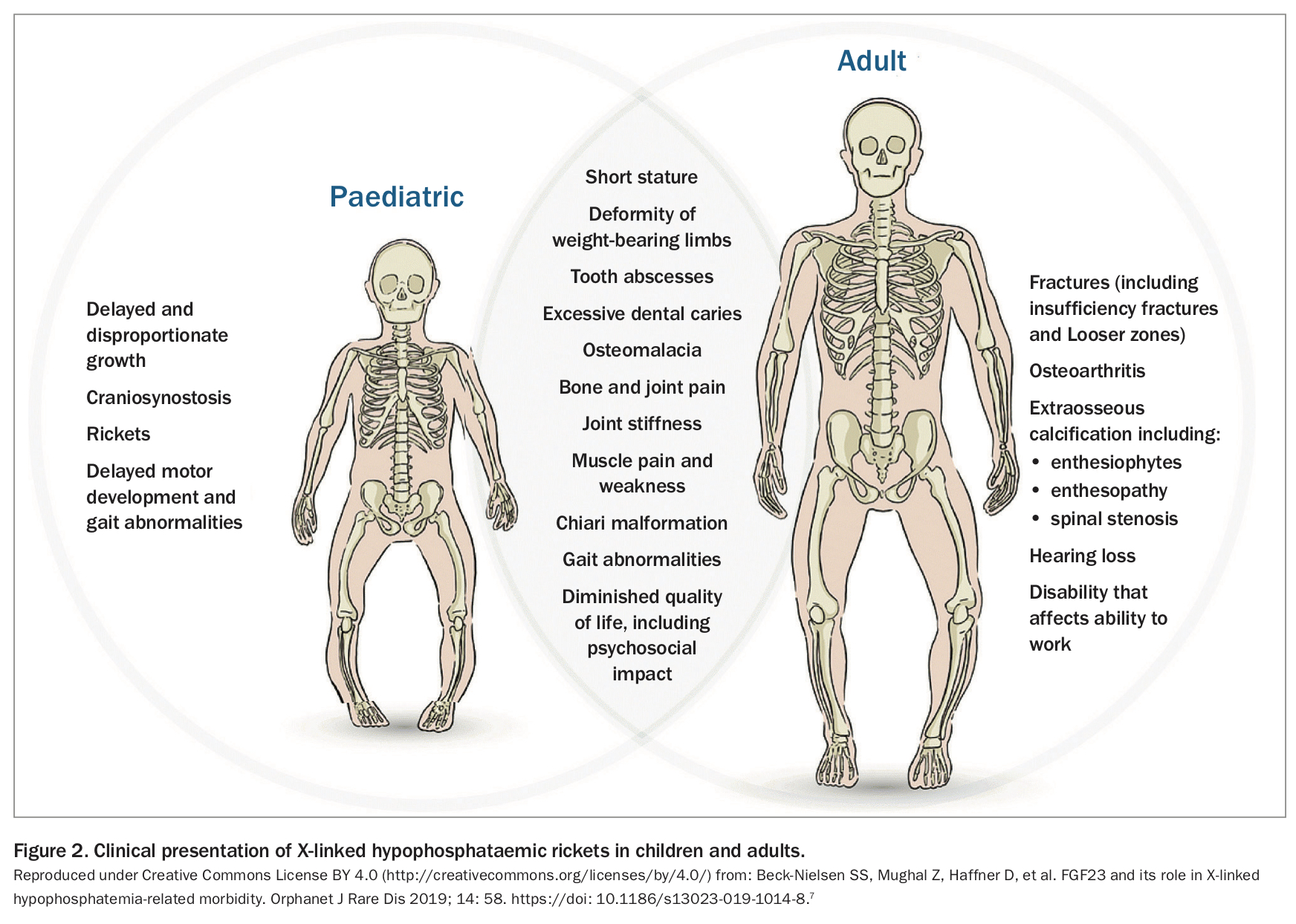{kind=link}
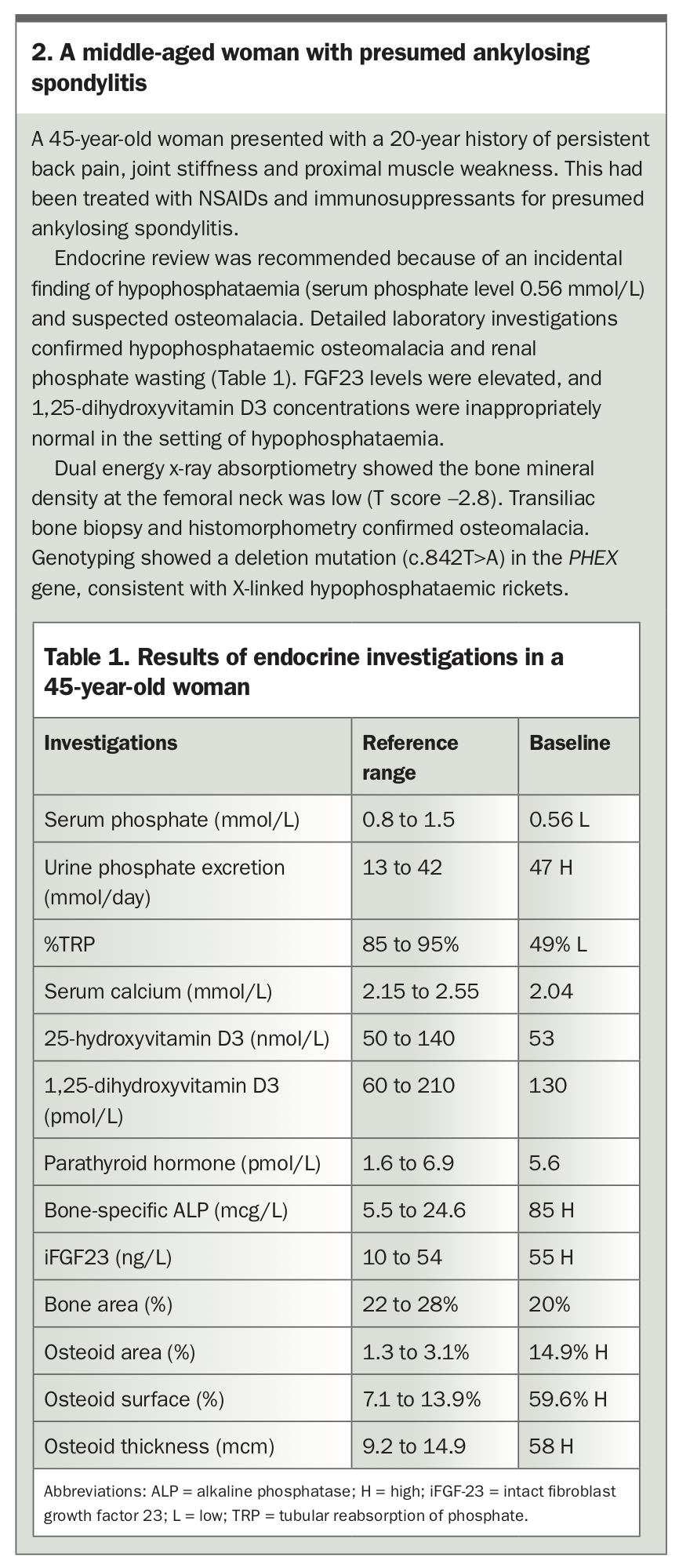{kind=link}
{kind=link}
Autosomal hypophosphataemic rickets
There are two forms of autosomal hypophosphataemic rickets: a dominant and a recessive form. Autosomal dominant hypophosphataemic rickets is caused by an activating mutation in the FGF23 gene at the proteolytic cleavage site, which renders FGF23 resistant to cleavage, increasing circulating levels.
Autosomal recessive hypophosphataemic rickets results from inactivating mutations in the DMP1 and FAM20C genes, which increase FGF23 expression and reduce FGF23 cleavage, respectively.
Acquired causes of FGF23-mediated hypophosphataemia
Tumour-induced osteomalacia
Tumour-induced osteomalacia is an acquired cause of hypophosphataemic osteomalacia caused by hypersecretion of FGF23, usually by benign neoplasms of mesenchymal origin. These include phosphaturic mesenchymal tumours, nonossifying fibromas, fibroangiomas and giant cell tumours.7
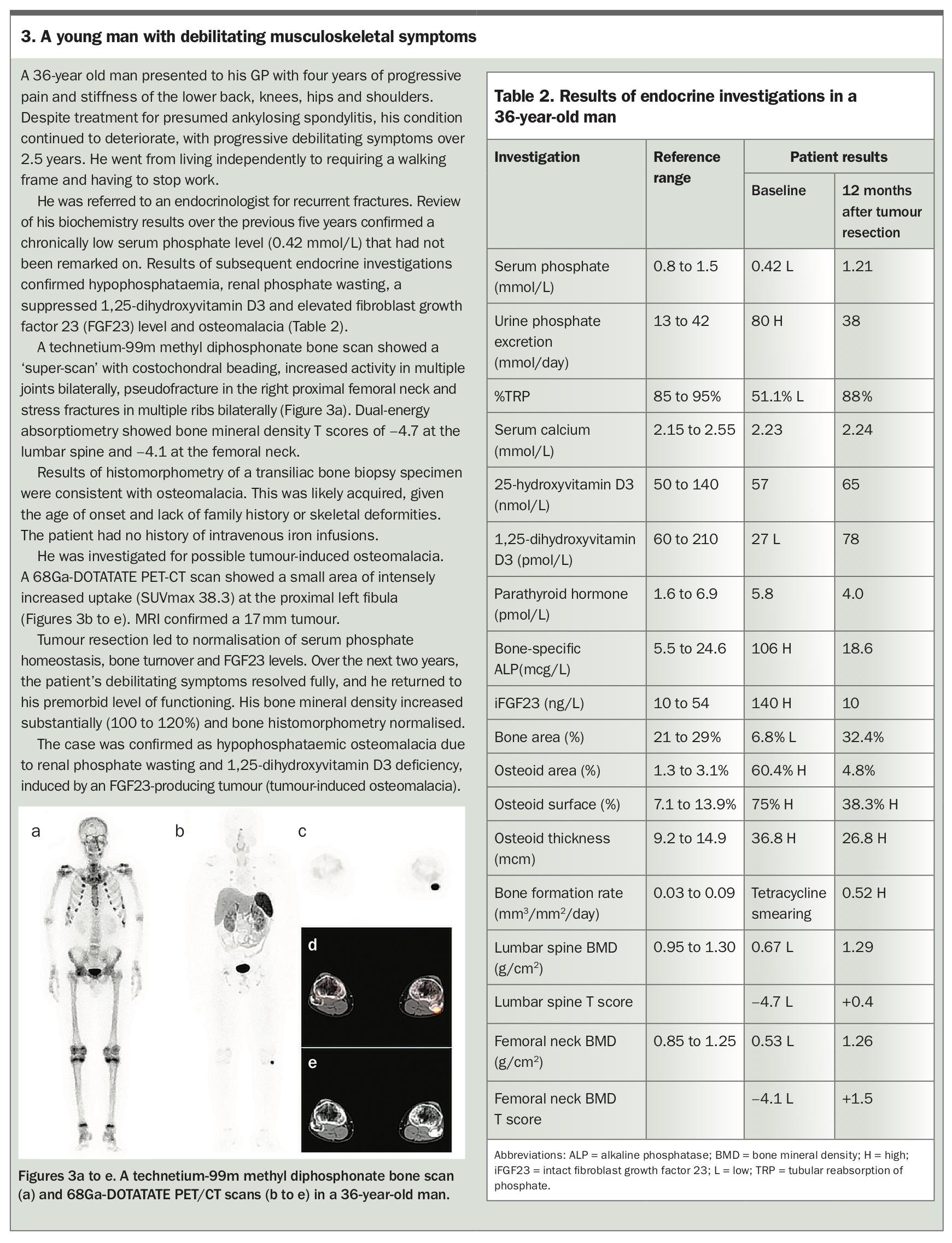
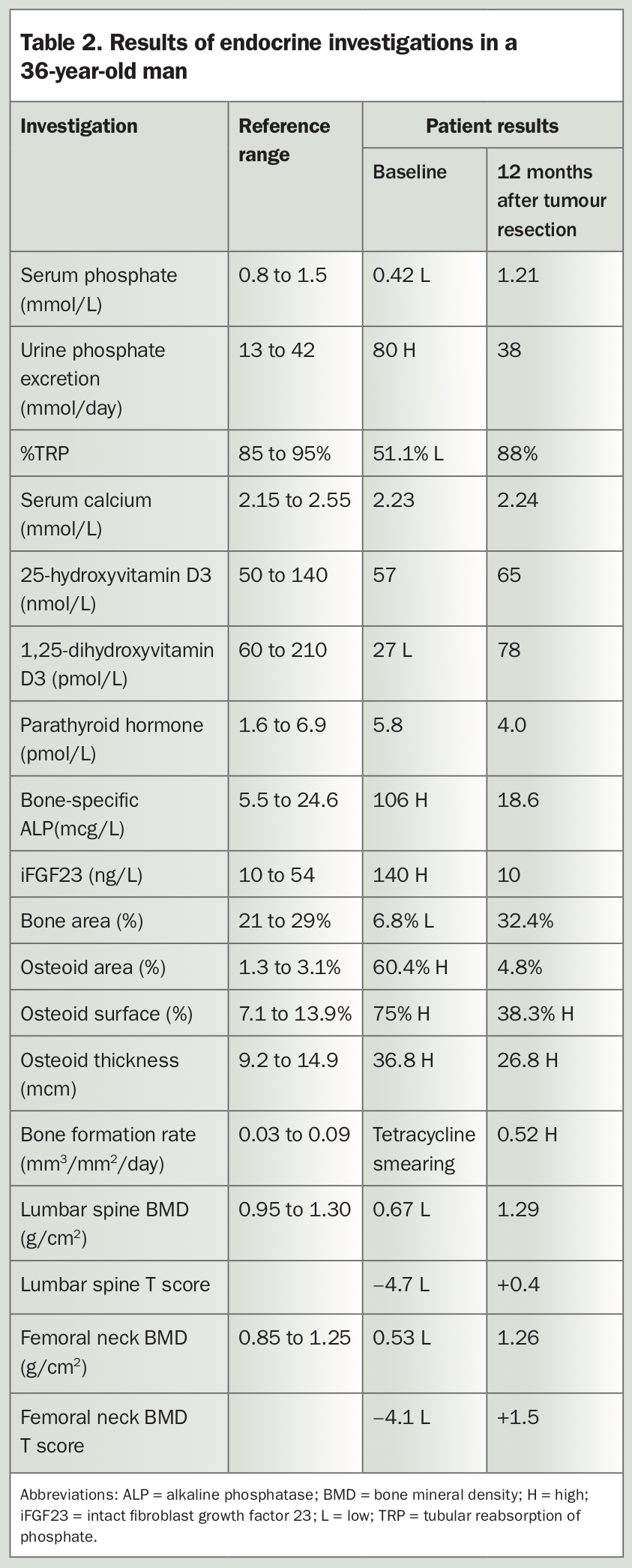
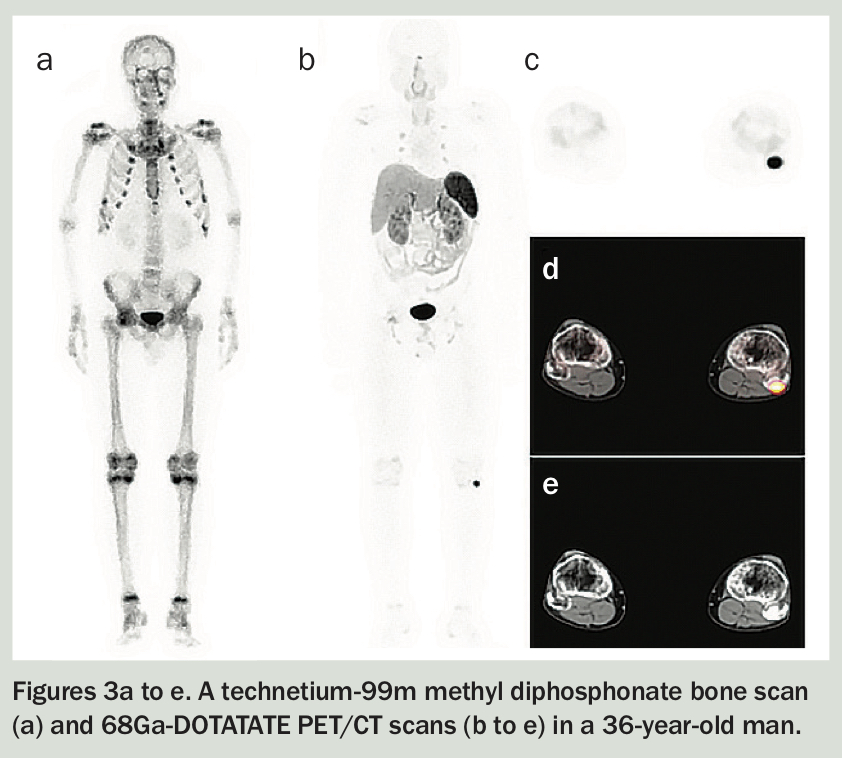
Phosphaturic mesenchymal tumours are often benign and slow-growing. The major cause of morbidity in patients with these tumours is delayed diagnosis of the paraneoplastic syndrome of hypophosphataemic osteomalacia, rather than tumour mass effect or metastases.8 A young man with a four-year history of progressive debilitating musculoskeletal symptoms due to tumour-induced osteomalacia is described in Box 3, Table 2 and Figures 3a to e.
{kind=link}
{kind=link}
{kind=link}
Iron infusion-associated osteomalacia
Treatment of iron deficiency with intravenous infusions of iron, particularly ferric carboxymaltose, has become an important and not-so-rare cause of hypophosphataemia. The pathogenic mechanisms remain uncertain. The first evidence linking iron infusion-associated hypophosphataemia to FGF23 excess arose from a prospective New Zealand study on single-dose intravenous iron polymaltose in eight female outpatients.9 More recent evidence indicates ferric carboxymaltose may reduce cleavage of FGF23.
Two identical randomised controlled trials of single-course iron infusions in adults with iron deficiency anaemia found the incidence of hypophosphataemia was significantly greater after ferric carboxymaltose (74.4%) compared with iron isomaltoside (8.0%) infusions.10 Ferric carboxymaltose resulted in increased circulating FGF23 concentrations within 24 hours of the infusion, and nadir TRP, serum phosphate and 1,25-dihydroxyvitamin D3 levels after two weeks, with hypophosphataemia persisting to six to 12 weeks.10
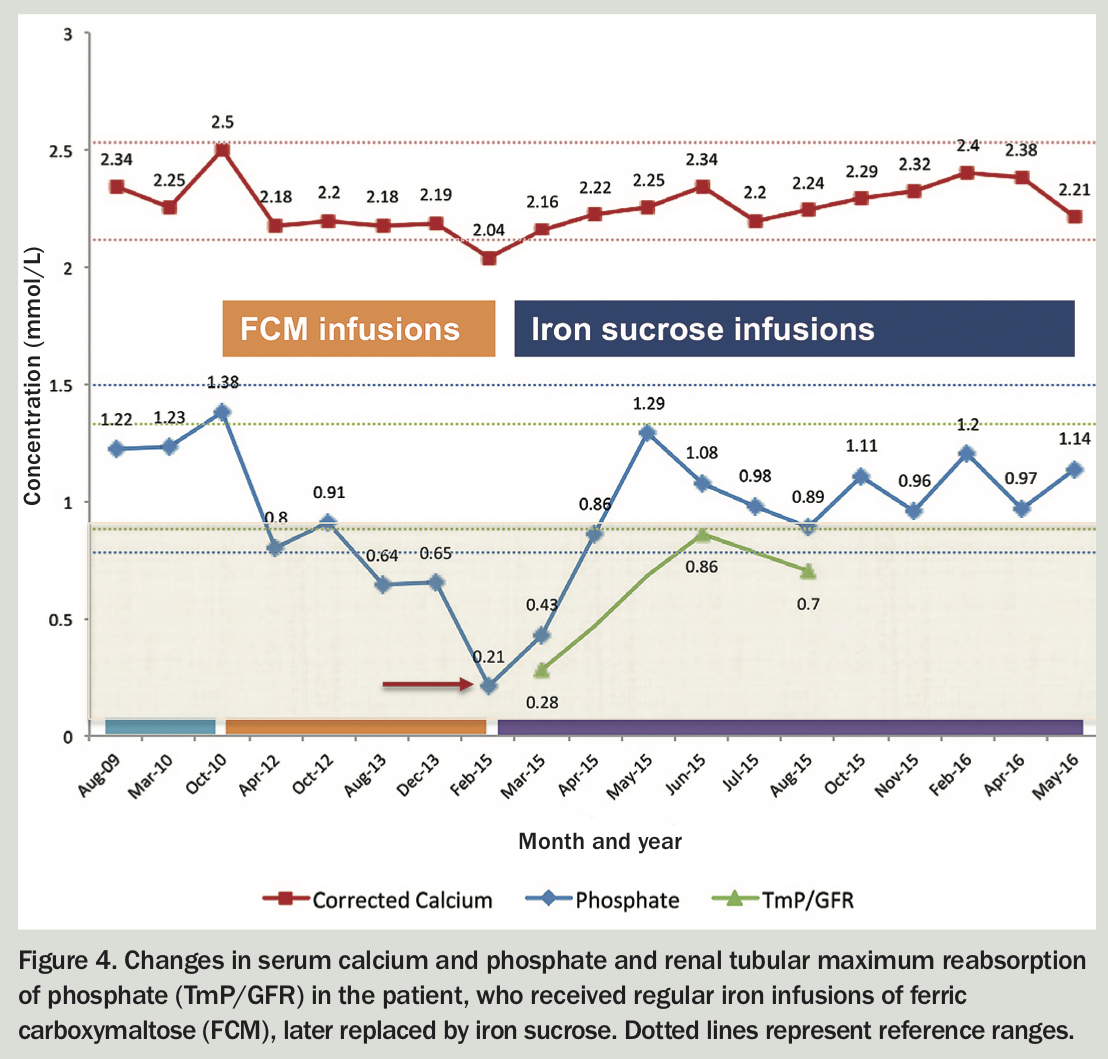
Randomised controlled trials indicate 50 to 70% of patients treated with ferric carboxymaltose develop incident hypophosphataemia. However, data on severe, persistent or symptomatic hypophosphataemia are scarce.10-12 A patient with hypophosphataemic osteomalacia associated with iron infusions is described in Box 4, Figure 4 and Table 3.
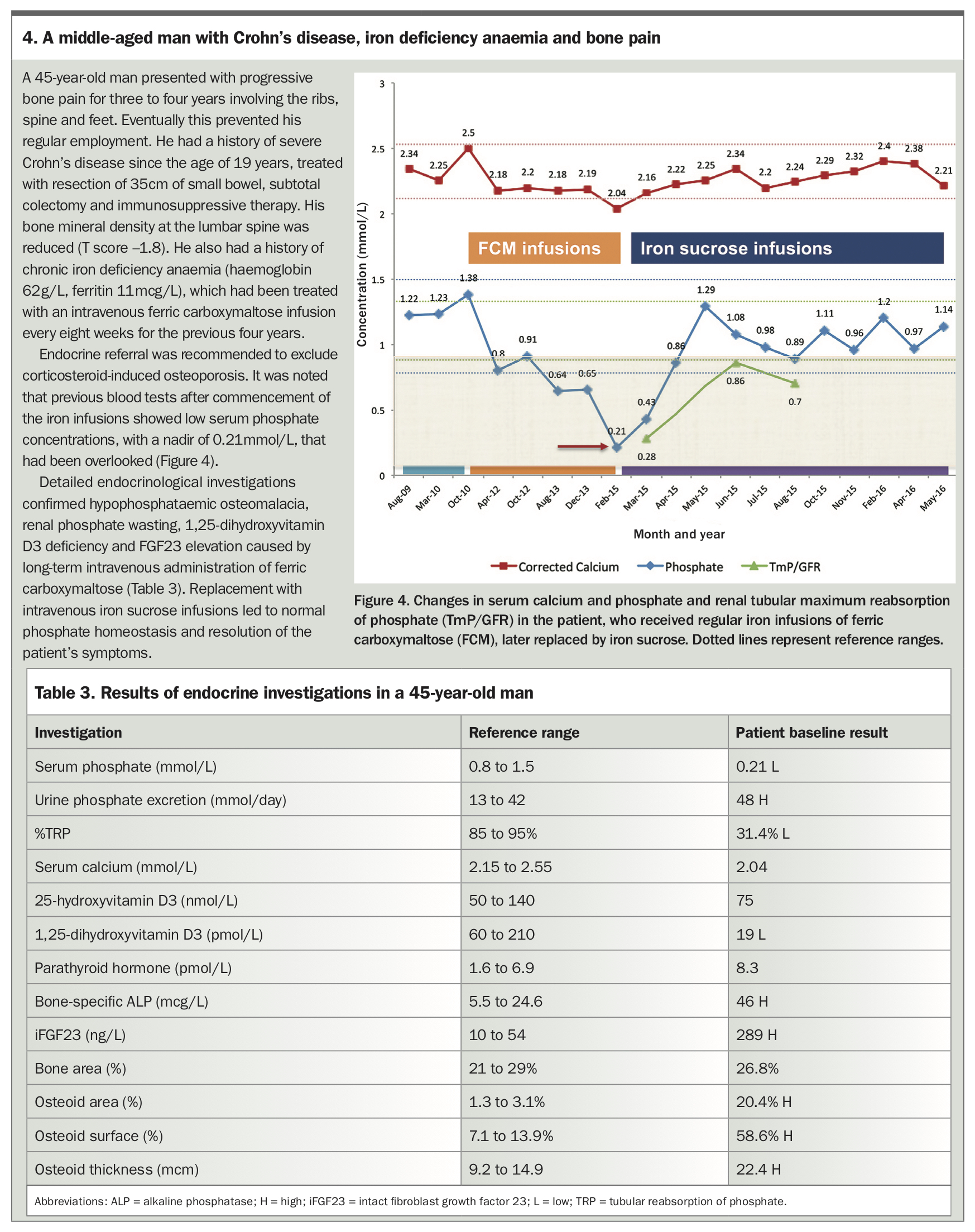{kind=link}
{kind=link}
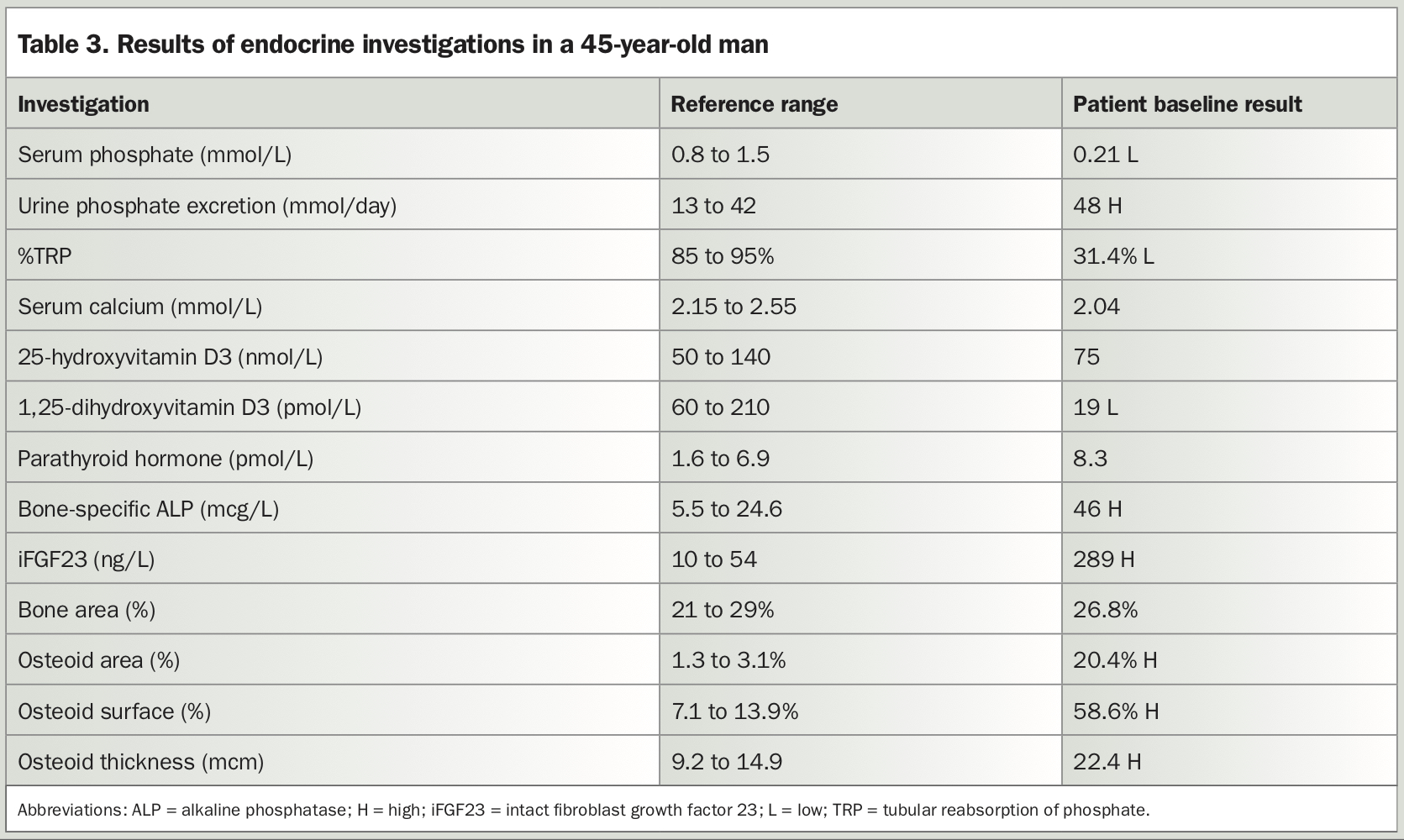{kind=link}
Non-FGF23-mediated causes of renal phosphate wasting and hypophosphataemia
Non-FGF23-mediated causes of renal phosphate wasting leading to hypophosphataemia include primary hyperparathyroidism, in which PTH increases renal phosphate excretion, primary and secondary renal tubular disorders (e.g. Fanconi syndrome and amyloidosis) and medications that affect the proximal convoluted tubule (e.g. ifosfamide, cisplatin and tenofovir).
Specialised investigations
FGF23 assay
Measurement of the FGF23 level is useful in determining whether renal phosphate wasting is FGF23-mediated. However, it is not crucial as serum hypophosphataemia associated with renal phosphate wasting and a low or inappropriately normal 1,25-dihydroxyvitamin D3 level is highly indicative of FGF23 excess.
Several FGF23 immunoassays are commercially available.13 The automated intact FGF23 assay (iFGF23) is used in Australian clinical practice but is not rebatable by Medicare. This chemiluminescence immunoassay measures intact 251 amino acid-length protein (iFGF23) by simultaneous recognition of epitopes on the N- and C-terminal domains. In healthy adults, the reference range for the plasma iFGF23 level is 10 to 54 ng/L. Blood samples for this test should be collected in EDTA tubes and centrifuged, and the EDTA plasma frozen at −20oC for transport to the laboratory.
The C-terminal FGF23 assay (cFGF23) detects both intact FGF23 and the inactive C-terminal fragment. However, this assay is not routinely used in Australia.
Bone biopsy
The skeletal complications of hypophosphataemia include recurrent stress fractures, low bone mineral density and hypomineralising bone disorders.14 A tetracycline-labelled bone biopsy is the ‘gold standard’ diagnostic tool to characterise osteomalacia and carefully evaluate the rate of bone mineralisation. Histomorphometric features consistent with osteomalacia include elevated osteoid area, surface and thickness (hyperosteoidosis). Although bone biopsy is highly specialised, not readily available and not necessary to make a diagnosis of osteomalacia, it can be useful in this clinical context.
Treatment of hypophosphataemia secondary to FGF23 excess
Management of chronic hypophosphataemia secondary to excess FGF23 (with or without rickets or osteomalacia) requires a multidisciplinary team approach, with input from endocrinologists, physiotherapists, occupational therapists, geneticists (if an inherited cause is suspected) and orthopaedic surgeons (to correct skeletal deformities or resect tumours in patients with tumour-induced osteomalacia).
Standard treatment involves oral phosphate replacement, which is often limited by the need for multiple daily doses and gastrointestinal intolerance. Calcitriol replacement is essential to correct 1,25-dihydroxyvitamin D3 deficiency, promote gastrointestinal phosphate absorption and prevent secondary or tertiary hyperparathyroidism. Treatment can be complicated by hypercalciuria and nephrocalcinosis.15,16
Recent randomised clinical trials have compared burosumab (an anti-FGF23 monoclonal antibody) versus placebo for adults and children with XLH and found significant improvements in phosphate homeostasis, symptoms, quality of life and fracture healing.17 Burosumab is also being trialled as a treatment for tumour-induced osteomalacia.18 It has been used successfully in a patient with iron infusion-associated hypophosphataemic osteomalacia.19
Burosumab received TGA approval in September 2021 for use in patients aged 1 year or older with XLH. However, it is not PBS-approved for this indication, and its clinical use in Australia is currently limited by cost and availability.
Conclusion
FGF23-mediated renal phosphate wasting and hypophosphataemia can present with features of osteomalacia, including bone pain, proximal muscle weakness, recurrent stress fractures, and mobility and functional decline. Measurement of the serum phosphate concentration is essential in the assessment of patients presenting with chronic musculoskeletal disorders to ensure timely diagnosis and management of this often overlooked, debilitating disorder. Recent discoveries about the genes that regulate FGF23 have led to better understanding of genetic disorders such as XLH and autosomal hypophosphataemic rickets. Excess circulating FGF23 is also responsible for the pathogenesis of tumour-induced osteomalacia and hypophosphataemia associated with iron (ferric carboxymaltose) infusions. The anti-FGF23 monoclonal antibody burosumab may have a role in management of XLH and other FGF23 excess syndromes but is currently limited by cost and availability. ET
COMPETING INTERESTS: None.