Medical management of acromegaly: a growth area
Growth disorders
Pituitary disorders
Acromegaly is a rare condition of the pituitary gland that may go undetected for many years because clinical presentations, particularly in adults, are subtle. Screening to detect an elevated insulin-like growth factor 1 level can confirm the diagnosis and surgery can offer the best chance of cure. When surgery is unsuccessful or contraindicated, a new generation of recently PBS-listed medications offer alternative effective treatment options to patients.
- Acromegaly is a rare condition, often associated with a significant delay in diagnosis.
- Uncontrolled acromegaly causes significant morbidity and an average 10-year reduction in life expectancy.
- Surgery is the recommended first-line treatment.
- A growing number of medical therapies are available for patients in whom surgery is unsuccessful or contraindicated, including somatostatin receptor ligands (octreotide, lanreotide, pasireotide), a dopamine agonist (cabergoline) and a newer growth hormone receptor antagonist (pegvisomant), which can achieve a hormonal response in up to 95% of patients.
Acromegaly is a rare disease caused by excessive production of growth hormone (GH), most commonly by a benign pituitary adenoma. Estimated prevalence varies widely from 50 to 1000 cases per million population. Because onset of symptoms may be subtle, a large number of cases are thought to go undiagnosed.1,2
GH stimulates production of insulin-like growth factor 1 (IGF-1) – primarily by the liver – which mediates its actions. Acromegaly is a multisystem disorder resulting from soft tissue overgrowth and metabolic derangements caused by GH and IGF-1 hypersecretion. Uncontrolled acromegaly is associated with reduced life expectancy due to elevated risk of cardiovascular disease.3 Successful treatment to normalise GH levels is critical to improve quality of life and restore lifespan.
Options for medical management of acromegaly have expanded with the PBS listing of the GH receptor antagonist pegvisomant and new generation somatostatin receptor ligand (SRL) pasireotide in recent years. In this article, we discuss how medical therapies fit into current acromegaly treatment guidelines.
Presentation of acromegaly
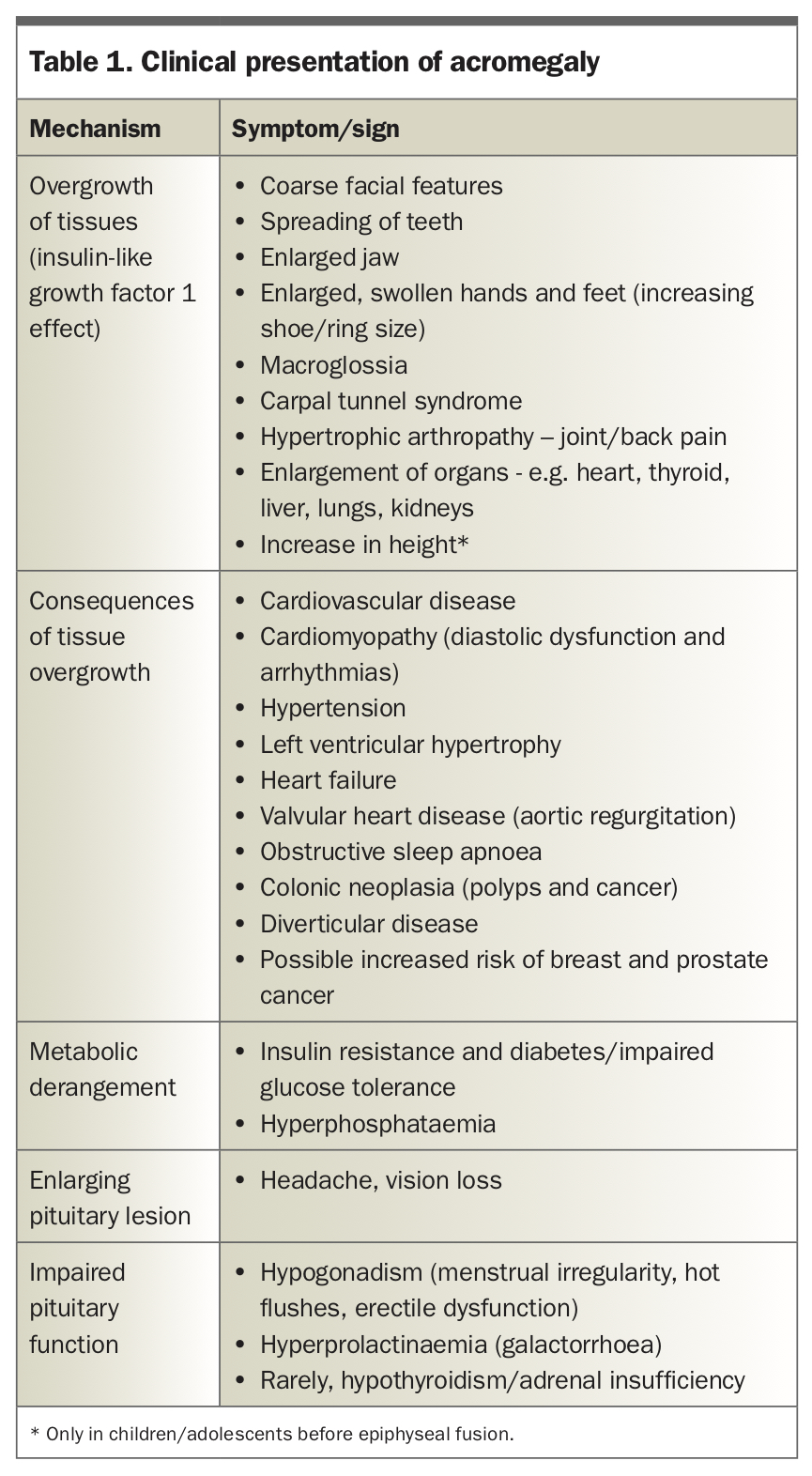
Acromegaly in children and teenagers, also known as pituitary gigantism, is associated with excess GH levels before the fusion of epiphyseal growth plates and results in a rapid increase in height. However, adults, who have reached skeletal maturity, will not grow taller with GH excess. Classical clinical manifestations in adults include prominence of brow, jaw and nose, spreading of teeth and increase in size of hands and feet (Table 1). Changes in appearance occur slowly and may not be noticed, and one in four patients has a delay in diagnosis of more than 10 years.4 Comparison with old photographs may reveal the gradual transformation of facial features over time.
{kind=link}
Macroglossia and soft palate enlargement can lead to obstructive sleep apnoea. Patients may develop carpal tunnel syndrome or back pain due to hypertrophic arthropathy.
Patients are at risk of hypertension and heart failure due to cardiomyopathy, as well as new or worsening diabetes due to a paradoxical hyperglycaemic effect of IGF-1. There is an increased risk of colonic polyps and diverticular disease, and possibly increased risk of breast and prostate cancer.
Patients may also present with visual loss due to compressive symptoms of a large pituitary lesion. Headaches are a common symptom in patients with acromegaly and have a complex pathogenic basis. Acromegaly can also present with symptoms of pituitary hormone deficiency, most commonly hypogonadism and, less frequently, central adrenal insufficiency or hypothyroidism.
Who to investigate
Patients who have clinical features of acromegaly and any patient with a pituitary adenoma should have their IGF-1 level measured. Although not every patient with diabetes or sleep apnoea needs to be screened, consider IGF-1 testing in patients presenting with a number of related conditions, such as sleep apnoea, uncontrolled diabetes mellitus, carpal tunnel syndrome, heart failure and colonic polyps – even without typical physical characteristics of acromegaly.5
Investigations
The best test for diagnosing acromegaly is measuring serum IGF-1 level, which remains stable across the day, unlike GH level, which fluctuates. A clearly elevated IGF-1 level confirms the diagnosis and has good rule-out value if negative.
Equivocal IGF-1 levels can be confirmed with an oral glucose tolerance test. This involves testing GH levels before and after a 75 g glucose drink (at 0 mins, 30 mins, 60 mins, 90 mins, 120 mins). High glucose intake normally suppresses GH levels below 1 mcg/L.5 A GH level above this threshold after glucose loading confirms a diagnosis of acromegaly.
Pituitary MRI can determine the presence of a pituitary lesion after biochemical acromegaly is established. Rarely, when the MRI is normal or suggests pituitary hyperplasia, the cause may be ectopic GH or GH-releasing hormone secretion by a neuroendocrine tumour.
Measurement of other pituitary hormone axes (e.g. prolactin, thyroid and adrenal) is important to diagnose hormonal cosecretion or hypopituitarism. Detection and management of complications (e.g. echocardiogram, sleep study, colonoscopy) is also essential in the work-up of a patient with acromegaly and in preparation for surgery.
Rarely, acromegaly can occur as part of a familial endocrine tumour syndrome. Patients aged under 30 years with a family history of pituitary tumours or with features of a related genetic condition (e.g. multiple endocrine neoplasia type 1, Carney complex or McCune-Albright syndrome) should be referred to a specialised genetics service.6
Treatment
Surgery
Transsphenoidal surgery is the recommended first-line treatment, as it gives the best chance of cure (Flowchart).7 Patients with smaller tumours (<1 cm, i.e. microadenomas) and no invasion into the cavernous sinus who are operated on by experienced pituitary neurosurgeons have the highest rates of cure (>90%), whereas patients with larger, invasive tumours have cure rates of around 40 to 60%.8
In patients who have large or invasive tumours that are unable to be fully resected, surgical debulking is still beneficial as it may improve response to other treatment modalities and reduce compression on critical structures such as the optic chiasm.5
GH levels are rechecked after surgery, sometimes with a postoperative oral glucose tolerance test. If GH levels remain nonsuppressible, this suggests incomplete resection, and a return to theatre may be needed, provided the residual tumour is readily accessible.
Although undetectable initial postoperative GH levels of below 1ng/mL indicate surgical success, IGF-1 levels may take up to three months to normalise. Therefore, formal reassessment of IGF-1 levels and pituitary MRI around 12 weeks after surgery is necessary to confirm hormonal response.
Medication
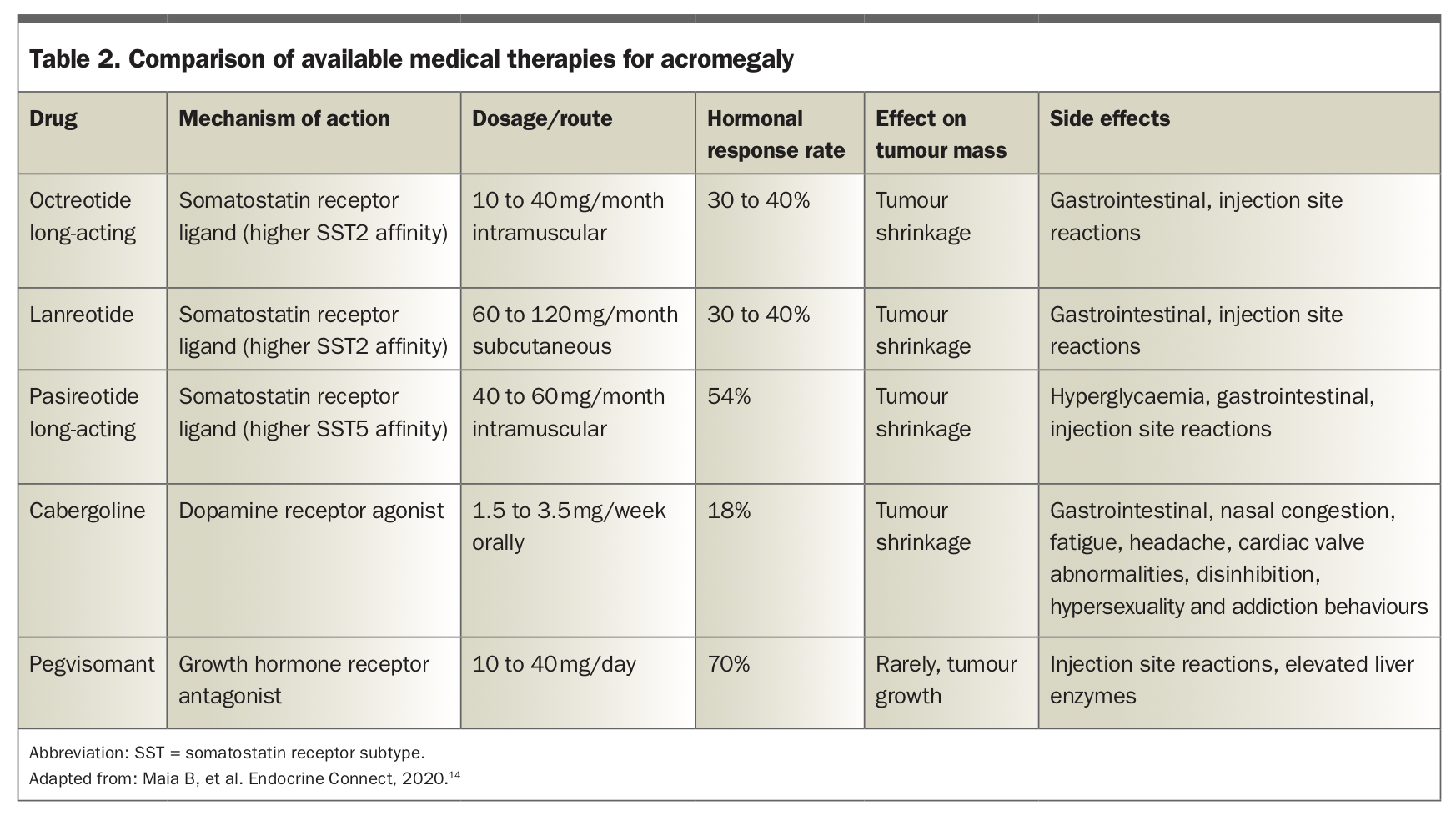
The options for medical treatment in patients for whom surgery is not curative, or is contraindicated, are summarised in Table 2.
{kind=link}
Somatostatin receptor ligands
Somatostatin is an inhibitor of GH secretion. SRLs that mimic its action are an important treatment for neuroendocrine tumours, including pituitary tumours that cause acromegaly. As well as reducing GH secretion, they can have anti-proliferative effects leading to tumour shrinkage of up to 20% in size, and are recommended as the first-line medical therapy in current guidelines.5
Octreotide and lanreotide are first-generation SRLs that act predominantly at somatostatin receptor type 2 sites. They are administered as either multiple daily injections or in a monthly long-acting form. Octreotide or lanreotide can achieve biochemical control in one in every two to three patients.
Pasireotide is a second-generation SRL with additional activity against the somatostatin type 5 receptor and is used as second-line therapy in resistant disease.7 It is also administered via monthly injection. The rates of biochemical control are slightly higher than with octreotide and lanreotide, but its antitumoural response rate is less than 50%.
The main side effect of SRLs is hyperglycaemia, with pasireotide having the highest rates, affecting up to 70% of cases. Less common adverse effects include gall stones, bradycardia and hair loss.
Dopamine agonist
Cabergoline, most commonly used as a treatment for prolactin-secreting pituitary lesions, can have a beneficial inhibitory effect on acromegaly by mimicking dopamine action – although fewer than one in three cases respond.9,10 The advantage of cabergoline is that it is an inexpensive oral tablet and, therefore, may be trialled in patients who have only mild elevations of IGF-1 (<2.5 times the upper limit of normal).
Growth hormone receptor antagonist
Pegvisomant is a novel second-line therapy, PBS listed in late 2016 for patients who have a persistently elevated IGF-1 level despite high-dose SRL. As a GH receptor antagonist, pegvisomant blocks the action of GH and reduces the production of IGF-1, thereby reducing the end-organ effects of GH excess. GH levels may paradoxically rise (due to blocking of IGF-1 feedback on the pituitary) and are not a guide to its efficacy. Although initial data using pegvisomant showed impressive rates of IGF-1 normalisation in up to 95% of patients, subsequent real-world experience indicates lower, though still impressive, response rates of around 70%.11,12 Disadvantages include administration via daily injection and a very small risk of hepatotoxicity requiring monitoring of liver function.
Unlike the other available treatments, pegvisomant does not act on the pituitary lesion itself and, rarely, may promote tumour growth. Therefore, regular monitoring with pituitary MRI will be needed to detect tumour progression.
Future directions
Combinations of SRL/pegvisomant have been trialled to increase the repertoire of therapies and hence the likelihood of successfully treating acromegaly and mitigating the effects of pegvisomant on tumour progression, although current PBS restrictions only allow for the use of one agent at a time.13
An oral formulation of octreotide has recently been approved by the US Food and Drug Administration and may become available in Australia in the future. Additional trials of oral somatostatin receptor and antisense oligonucleotide (mRNA-blocking) GH receptor inhibitors are under investigation. Improvement in biomarker analysis may lead to individualised treatment regimens.14
Radiotherapy
Stereotactic radiotherapy is currently reserved for patients in whom surgery and medical therapy have failed. The maximal effect of radiotherapy may only be seen after five to 15 years, necessitating continued medical therapy as a bridge. SRLs should be interrupted during radiotherapy to optimise treatment effect. Remission rates are 50 to 70%; however, the main limitation is a high burden of side effects such as hypopituitarism (15 to 40%), visual deterioration (1 to 3%), cognitive change, cerebrovascular disease, cranial nerve palsy and secondary tumours such as meningioma.15 Onset of these complications may be delayed, so long-term monitoring is important.
Gamma Knife Radiosurgery is a new technique that may shorten the interval to remission and reduce complication rates; however, data on its long term success are lacking.
Conclusion
Acromegaly causes significant morbidity and mortality driven by cardiovascular disease and may go undetected for many years. Screening for an elevated IGF-1 level should be performed in patients with suspicious physical features, pituitary lesions, or multiple, potentially related conditions. Recent advances in medical treatment allow patients with persistent acromegaly after surgery to achieve control of IGF-1 and GH levels without requiring radiotherapy or if radiotherapy is unsuccessful (see Case study in the Box). Reassuringly, patients with controlled acromegaly have similar mortality rates to the general population and improved quality of life. ET
{kind=link}
COMPETING INTERESTS: None.