Failure to thrive in children: when the cause is due to hormones
Growth disorders
Child development
Assessment of failure to thrive (FTT) in children can be challenging and early consultation with a general paediatrician is counselled. Although endocrine causes are rare, the consequences of missing them can be life-threatening and they are relatively easily excluded. This article presents a series of cases and screening investigations to help direct investigation or appropriate referral for different endocrine disorders that may present as FTT.
- Failure to thrive describes poor weight gain with preserved linear growth, although linear growth failure will follow in time.
- Most endocrine causes of failure to thrive are uncommon but important to consider and screen for.
- The key to excluding endocrine causes is to think of them.
Failure to thrive (FTT) is a description rather than a diagnosis,1, 2 referring to poor weight gain resulting in the downward crossing of percentiles associated with a relative sparing of linear growth. In infancy, this is not necessarily pathological and may represent either reversion to the mean of a big baby or catch-down to genetic potential. Although organic or psychosocial pathology is identified in fewer than 85% of cases of FTT,2 it needs to be excluded, particularly when poor weight gain is prolonged (e.g. more than six to 12 weeks in an infant) and/or out of keeping with the family background.
Initial assessment
In the first instance, assessment of a child with FTT should include the gathering of past growth data, including birth parameters, parental and sibling stature and growth history, and careful evaluation of the child’s nutritional intake and psychosocial wellbeing. Accurate measurement of the child’s length or height, weight and head circumference using a reproducible technique is important for comparison. Measurements should be plotted on appropriate growth charts and corrected for significant prematurity (i.e. if the baby is more than four weeks premature, subtract the number of weeks of prematurity from the postnatal age when plotting growth parameters in the first two years of life). The infant’s nappy should be removed because it makes the weight unreliable and interferes with accurate measurement of supine length. Supine lengths are used until the child will reliably stand for a measurement and hence growth charts are based on lengths up to 3 years of age. Supine length exceeds standing height by up to 0.7 cm.
The period of observation before initiating paediatrician referral depends on the presence of other red flags such as:
• rapid unexplained weight loss
• irritability
• vomiting
• symptoms suggestive of hypoglycaemia (e.g. lethargy, pallor, sweating, not waking for feeds)
• polyuria (frequent soaked nappies or secondary enuresis in an older child) and polydipsia (increased drinking through the day or overnight and a preference for water)
• abnormal examination findings such as decreased subcutaneous fat (saggy buttocks), hyperpigmentation of skin or gums, dysmorphism, neurological abnormality or hepatomegaly, developmental delay or suspicion of abuse or neglect.
Prolonged failure to gain weight will eventually lead to failure of linear growth, especially in infants. If linear growth crosses percentiles at the same time as weight, so that the child’s weight for height is preserved, this might represent either catch-down to genetic potential or point to a problem with linear growth for which endocrine aetiologies need to be excluded.
Causes of FTT
The possible causes of FTT can be categorised as the following:
• inadequate nutrient intake
• excessive nutrient loss
• metabolic requirements exceeding intake creating a net catabolic state.
By far the most common cause of FTT is inadequate nutrient intake. An endocrine aetiology for FTT is rare, even in a selected population of children with FTT referred to a paediatric endocrinology clinic. As shown in the case presentations, common findings associated with FTT are the irritable baby who feeds poorly and vomits, which are certainly not specific for an endocrine cause.
Once FTT is established in the primary care setting, early discussion with or referral to a paediatrician is suggested. Given the many caveats to endocrine investigations and their interpretation, early discussion with a paediatric endocrinologist for suspected hormonal causes of FTT may be beneficial.
The major endocrine disorders that may be associated with FTT and screening investigations for them are discussed below.
FTT associated with inadequate intake
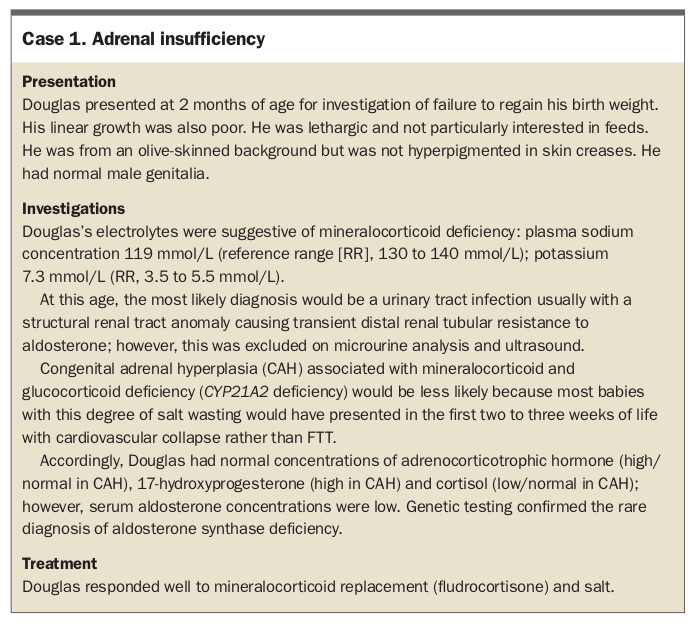
Adrenal insufficiency
Children with adrenal insufficiency tend to be prone to more severe and prolonged trivial illnesses than other members of their family. They have poor appetite and poor weight gain or even weight loss, but preserved linear growth. Adrenal insufficiency can be primary (i.e. the problem lies in the adrenal glands), associated with impaired ability of the adrenal glands to respond to adrenocorticotrophic hormone (ACTH), or secondary (i.e. the problem lies in the pituitary gland or hypothalamus), associated with ACTH deficiency. Secondary adrenal insufficiency is associated with glucocorticoid (cortisol) but not mineralocorticoid deficiency, because aldosterone production is independent of ACTH.
Irrespective of whether the adrenal insufficiency is primary or secondary, glucocorticoid deficiency is associated with a risk of hypoglycaemia (blood glucose level [BGL] below 3 mmol/L). In the absence of a documented low BGL, there may be a suggestive history of lethargy, not waking for feeds, pallor or sweating.
The clinical and biochemical manifestations of primary and secondary adrenal insufficiency also differ. Hyperpigmentation of non-sun-exposed areas of the skin, gum mucosa, skin creases and scars is a hallmark of primary adrenal insufficiency (PAI), as shown in Figures 1a to c. Hyponatraemia and potentially life-threatening hyperkalaemia are features of PAI, whereas the hyponatraemia of secondary adrenal insufficiency (SAI) is usually mild and plasma concentrations of potassium remain normal. SAI is very rarely isolated and the clinical and biochemical manifestations will also reflect deficiencies of other pituitary hormones (see ‘Pituitary insufficiency’ below).
{kind=link}
FTT is a prominent feature of isolated mineralocorticoid deficiency (hypoaldosteronism; Case 1) or, more often, aldosterone resistance (pseudohypoaldosteronism). It may also be associated with several monogenic, autoimmune and syndromic causes of PAI, but other presentations tend to predominate in these cases.2 This is illustrated by the most common cause of PAI in childhood, congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency (autosomal recessive CYP21A2 mutations; incidence one in 10,000 to 20,000), an enzyme crucial for cortisol and aldosterone synthesis.3 In the most severe form of CAH with less than 2% residual 21-hydroxylase activity causing severe glucocorticoid and mineralocorticoid deficiency, girls are usually diagnosed soon after birth because of the associated virilised genitalia. Diagnosis in boys is likely to be missed at birth; they will present with poor feeding, vomiting, lethargy, FTT, hyperpigmentation and various degrees of cardiovascular collapse due to adrenal crisis in the first two to three weeks of life. Milder degrees of deficiency present with early virilisation in infancy or early childhood (2 to 5% residual activity), or, in the late-onset form, with early pubic hair or acne in late childhood, or amenorrhoea in adolescent girls. Newborn screening for CYP21A2 deficiency was recently introduced in Australia, with the hope of reducing morbidity and mortality by early diagnosis.
{kind=link}
Investigations to screen for suspected PAI and SAI are listed in Box 1.
{kind=link}
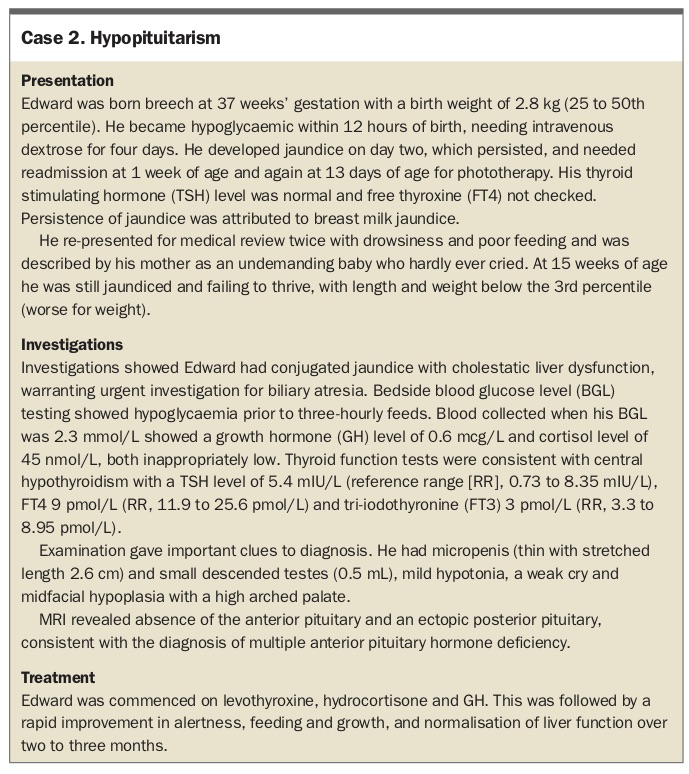
Hypopituitarism
FTT is a common association of undiagnosed congenital hypopituitarism. Because postnatal growth in the first six months of life is largely independent of growth hormone (GH), failure to gain weight may precede failure of linear growth even in infants with growth hormone deficiency. Birth parameters and gestational age are usually normal in affected infants, although there is an association with prolonged gestation and breech presentation.
The infant with hypopituitarism may be less active and not wake for feeds. The first few days of life may be marked by a history of apnoea, episodes of pallor or cyanosis, poor feeding and jitteriness, and nonspecific symptoms of hypoglycaemia that are sometimes mistaken (and treated) as sepsis. Prolonged conjugated or unconjugated jaundice in neonates is common (Case 2); however, liver impairment is fully reversible if pituitary hormone replacement is initiated in infancy.5 In male infants, micropenis offers a clue to the diagnosis. Some infants have temperature instability. Septo-optic dysplasia is associated with congenital hypopituitarism in 50% of cases,6 and this may be associated with congenital nystagmus and visual impairment.
{kind=link}
Early diagnosis and hormone replacement with GH, hydrocortisone and levothyroxine are crucial to avoid the deleterious effects on neurological development of hypoglycaemia and hypothyroidism.
Acquired causes of hypopituitarism that may present with FTT include suprasellar tumours such as craniopharyngioma, astrocytoma and dysgerminoma, and infiltrative causes such as Langerhans cell histiocytosis. The latter, in particular, may present with diabetes insipidus (see below).
Investigations to screen for suspected hypopituitarism are listed in Box 2. The length and weight charts of a girl diagnosed with hypopituitarism at age 2 years and 7 months are shown in Figures 2a and b.
{kind=link}
{kind=link}
Diabetes insipidus
Diabetes insipidus (DI) is caused by either inadequate vasopressin secretion by the posterior pituitary (central DI) or impaired action in the renal distal convoluted tubule (nephrogenic DI; see Case 3). Central DI can be isolated or associated with multiple pituitary deficiencies, trauma or an infiltrative or destructive process (the most common cause in childhood), or it may be genetic (autosomal dominant). Autosomal dominant central DI usually only starts to manifest in the second or third year of life. Congenital nephrogenic DI is usually X-linked but there are also autosomal recessive and (more rarely) dominant forms.
{kind=link}
Irrespective of the cause and type, DI results in obligatory urinary water loss and, as a consequence, increased thirst. Affected individuals have a strong preference for iced water. In hereditary forms, there may be a family history of storing litres of water in the refrigerator or taking many bottles on family outings. The affected child will have a history of frequent soaked and overflowing nappies or primary or secondary enuresis. A mobile child with an intact thirst mechanism will seek water at the expense of either calorie-rich fluids such as milk, or food intake, causing FTT (as in Case 3). Renal sodium retention in an effort to maximise fluid retention results in an inappropriately low (often undetectable) urinary sodium for the plasma sodium; nevertheless, plasma sodium can be normal if the child is an efficient drinker.
Screening investigations for suspected DI are listed in Box 3.
{kind=link}
Hypercalcaemia
Hypercalcaemia has protean manifestations with anxiety and mood disturbance, nonspecific aches and pains, polyuria, polydipsia and anorexia or fussy feeding causing poor weight gain. The affected infant is miserable and feeds poorly, causing FTT (see Case 4). End-organ damage due to hypercalcaemia (nephrocalcinosis, fractures or osteoporosis) by the time of diagnosis is not uncommon in children and adolescents, as the nonspecific symptoms and signs such as poor weight gain, easy fatigability, depressed mood and aches and pains are easily missed.
{kind=link}
The causes of hypercalcaemia can be divided into parathyroid hormone (PTH)- mediated and non-PTH mediated, with underlying aetiologies being different in neonates and infants compared with those in older children.
The hypercalcaemia mediated by PTH is usually severe. Neonatal severe hyperparathyroidism is, thankfully, rare, and due either to an adaptation in utero to maternal hypocalcaemia (in which case it is transient) or to homozygous inactivation of the calcium-sensing receptor (CaSR), resulting in unregulated secretion of PTH.7 In its milder form, an inactivating mutation of the CaSR gene is more often associated with familial hypocalciuric hypercalcaemia (FHH; autosomal dominant); however, most individuals with this are asymptomatic.7 Hyperparathyroidism in older children is usually sporadic (in more than 65% of cases), associated with a single parathyroid adenoma.7 It can also be the presenting feature of the autosomal dominant multiple endocrine neoplasia (MEN) syndromes, more often type 1 than type 2, in which case there may be a family history of nephrolithiasis, renal stones or parathyroidectomy.
More often, particularly in infancy, hypercalcaemia is not mediated by PTH and a cause can be hard to pin down, hence the term, idiopathic hypercalcaemia of infancy. This may be found incidentally, or when investigating for poor feeding, irritability and FTT. Excessive vitamin D supplementation or inherited defects in vitamin D metabolism,8,9 and excessive calcium supplementation in total parenteral nutrition (TPN) or in milk fortifiers for small or premature infants, need to be excluded. Subcutaneous fat necrosis of the newborn, which may manifest as faint pink macules or the feel of subcutaneous nodules or plaques, is associated with a history of birth trauma or asphyxia. Syndromic causes such as Williams syndrome (Case 4) and various rare inborn errors of metabolism can be associated with hypercalcaemia.10
In older children, prolonged immobility, especially in association with fractures, causes transient hypercalcaemia.7 Hypercalcaemia of malignancy is rare in children and mostly associated with haematological malignancies. Chronic vitamin A intoxication leads to hypercalcaemia, which also has been reported in association with vitamin A analogues used as chemotherapy for certain cancers.
Investigations to screen for hypercalcaemia are listed in Box 4.
{kind=link}
FTT despite normal appetite
Diencephalic syndrome
Diencephalic syndrome is rare and characterised by normal linear growth and cachexia despite good appetite in a hyperalert infant. It is most often associated with large hypothalamic or optic pathway gliomas, but not necessarily detectable hormonal abnormality (Case 5).11,12 Decreased subcutaneous fat contributes to the emaciated appearance of infants with diencephalic syndrome, but investigations do not show any micronutrient deficiencies otherwise associated with decreased nutritional intake.
{kind=link}
The cause of the FTT despite a good appetite has not been elucidated. Theories on its pathogenesis include increased GH levels, partial GH resistance, increased lipolysis and increased energy expenditure.
Diagnostic screening for diencephalic syndrome includes the investigations listed for hypopituitarism in Box 2, as well as a cerebral MRI.
Psychosocial dwarfism
The spectrum of psychosocial dwarfism is rather more common than diencephalic syndrome but usually associated with linear growth failure rather than poor weight gain, so strictly speaking is not a form of FTT. Appetite can be voracious but affected infants often have abnormal eating behaviour and vomiting in addition to other behavioural disturbance. Impaired emotional-cognitive development is common. In its extreme form, there are identifiable pituitary hormone abnormalities including central (pituitary) hypothyroidism, low insulin-like growth factor-1 (IGF-1) and GH deficiency on dynamic testing.
Psychosocial dwarfism is thought to be mediated by the hypothalamic effects of chronic stress.13,14 Growth does not respond to GH treatment but all pituitary abnormalities resolve with resolution of the adverse social circumstances.13
The diagnosis of psychosocial dwarfism is based on suggestive history and, importantly, on establishing reversal of growth failure after removal of the psychosocial stressor. Hypopituitarism must be excluded with screening as suggested in Box 2.
FTT associated with increased metabolic need
Diabetes mellitus
The main endocrine aetiology in FTT associated with increased metabolic need is type 1 diabetes mellitus, most often due to autoimmune destruction of the insulin-producing islet cells. Diabetes can develop at any age, including in neonates and infants, in whom a monogenic cause is more likely; however, until a specific genetic cause is established, insulin remains the appropriate treatment.
Without the anabolic action of insulin, glucose is lost in the urine and the net catabolic effect leads to weight loss in a child who is eating and drinking voraciously. The absence of insulin allows uncontrolled ketogenesis, leading to life-threatening acidosis and metabolic derangement, so this is an important diagnosis to not delay or miss. As shown in Case 6, the diagnosis may be delayed with life-threatening consequences when there are other plausible causes of weight loss such as psychiatric comorbidity or developmental delay.
{kind=link}
The key to making the diagnosis is to consider it. The next step is a urine dipstick for glucosuria (and ketonuria) or a capillary (finger-prick) blood glucose test and, if possible, measurement of ketones. These should be performed in the office.
Investigations to screen for diabetes mellitus are listed in Box 5.
{kind=link}
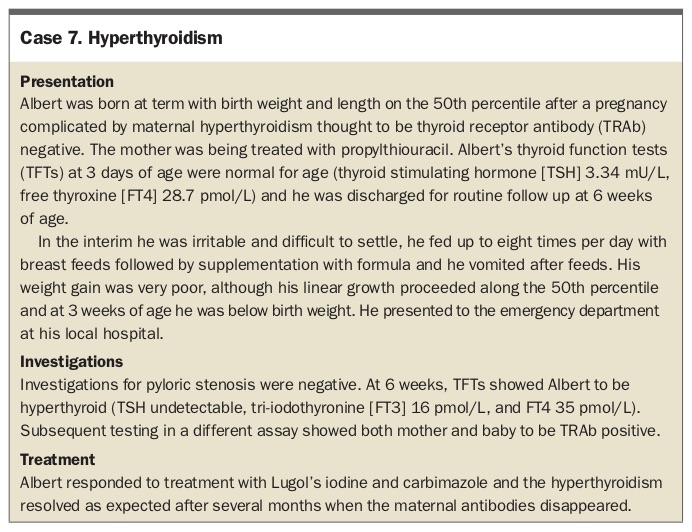
Hyperthyroidism
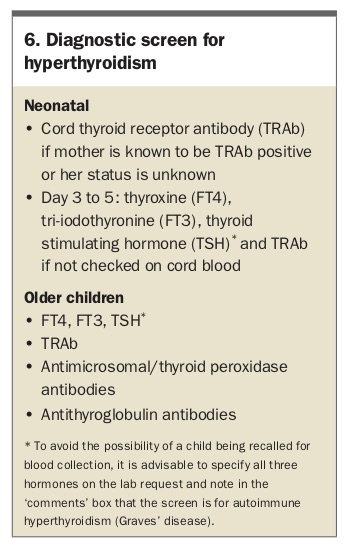
Hyperthyroidism at any age is associated with weight loss or poor weight gain despite a big appetite, because of the increased metabolic rate. The most common symptoms described beyond infancy are anxiety, restlessness, poor sleep, weakness and decreased exercise tolerance. Signs elicited on examination include tachycardia, warm sweaty hands, fine tremor, rapid correcting reflexes, exophthalmos, lid-lag and conjunctival chemosis. The neonate presents with poor weight gain, irritability, jitteriness and tachycardia. The most common aetiology is autoimmune thyroid disease, with hyperthyroidism caused by antibodies (thyrotropin-receptor antibodies; TRAb) stimulating the thyroid stimulating hormone (TSH) receptor.
Neonatal hyperthyroidism is usually due to transplacental passage of TRAb from mother to fetus. There should be a history of maternal hyperthyroid autoimmune thyroid disease (Graves’ disease). The onset of neonatal hyperthyroidism may be delayed when the mother is taking significant doses of antithyroid medications as these also cross the placenta and affect the newborn’s thyroid; hence infant review about the age of day 5 with repeat thyroid function testing is crucial.
Rarely, hyperthyroidism is due to an activating mutation of the TSH receptor. Inheritance is autosomal dominant (but it can be sporadic), so there may be a family history, but, importantly, the timing of onset is variable, even within kindreds.
Although modern TRAb assays have very high sensitivity and specificity, they are not perfect,16 and infant follow up should be guided by the clinical history, not just serological findings to avoid late diagnosis, as shown in Case 7. Missed neonatal thyrotoxicosis can have irreversible consequences including heart failure and craniosynostosis.
{kind=link}
TRAb-positive neonatal thyrotoxicosis resolves once maternal antibodies disappear from the infant’s circulation within a few months.
The diagnostic screening investigations for suspected hyperthyroidism are listed in Box 6.
{kind=link}
Summary
In children of any age with FTT, general screening investigations including serum electrolytes, calcium, phosphate and glucose levels; liver function tests; and full thyroid function tests, together with a good history and examination, will provide a reasonable indication of whether an endocrine abnormality is likely.
There are many caveats to the interpretation of endocrine investigations and if an endocrine cause is thought likely, early consultation with an endocrinologist is advisable. On the other hand, because most children will have another explanation for their poor weight gain, consultation with a general paediatrician may be of most help in the first instance. ET