Investigating polydipsia and polyuria

Urine and urination
Fluid and electrolyte balance
The polyuria-polydipsia syndrome includes three different conditions: central diabetes insipidus, nephrogenic diabetes insipidus and primary polydipsia. Differentiating between these entities is crucial but can been challenging in clinical practice. Recently, measurement of plasma copeptin, the C-terminal segment of the arginine vasopressin prohormone, has become routinely available and now has an important role in the differentiation of polyuria-polydipsia syndrome.
- Polyuria-polydipsia syndrome comprises central diabetes insipidus (DI), nephrogenic DI and primary polydipsia.
- The introduction of plasma copeptin measurements has advanced the investigation of polyuria-polydipsia syndrome.
- Baseline plasma copeptin measurements are useful for the diagnosis of nephrogenic DI.
- Stimulated plasma copeptin measurements are useful for the differentiation of central DI and primary polydipsia.
- The hypertonic saline-stimulated copeptin test has replaced the water deprivation test as the gold standard test for the diagnosis of central DI.
Polyuria is defined as the excretion of abnormally large volumes of dilute urine (>3 L/24 h or >40–50 mL/kg/24 h).1 The polyuria-polydipsia syndrome comprises three major conditions: primary polydipsia, central diabetes insipidus (DI) and nephrogenic DI.1,2 Differentiating between these entities accurately is crucial, as treatment approaches differ and inadequate or incorrect treatment may lead to severe complications. However, determining the diagnosis can be challenging, particularly when distinguishing primary polydipsia from partial, milder forms of central DI.
The traditional water deprivation test, previously the gold standard for the diagnosis of DI, has several limitations and poor diagnostic accuracy.3 Direct measurement of arginine vasopressin (AVP) is technically challenging for many reasons, and consequently not useful clinically.4-7 In comparison, copeptin, the C-terminal segment of the AVP prohormone, is easily measured.8 Copeptin measurements are now routinely available and have an important role in the differentiation of polyuria-polydipsia syndrome. This article provides an overview on the investigation of polyuria-polydipsia, including the role of plasma copeptin measurements.
Physiology
AVP is a nine-amino-acid peptide derived from pro-AVP, a 164-amino-acid precursor protein consisting of a signal peptide, the AVP moiety, neurophysin 2, and copeptin, a 39-amino-acid peptide.9 Pro-AVP is synthesised in the paraventricular and supraoptic nuclei of magnocellular neurons in the hypothalamus.10 Post-translational processing separates neurophysin, AVP and copeptin during transport down the pituitary stalk to axon terminals in the posterior pituitary, where AVP is stored in neurosecretory granules until specific stimuli cause secretion into the circulation (Figure).2,10,11
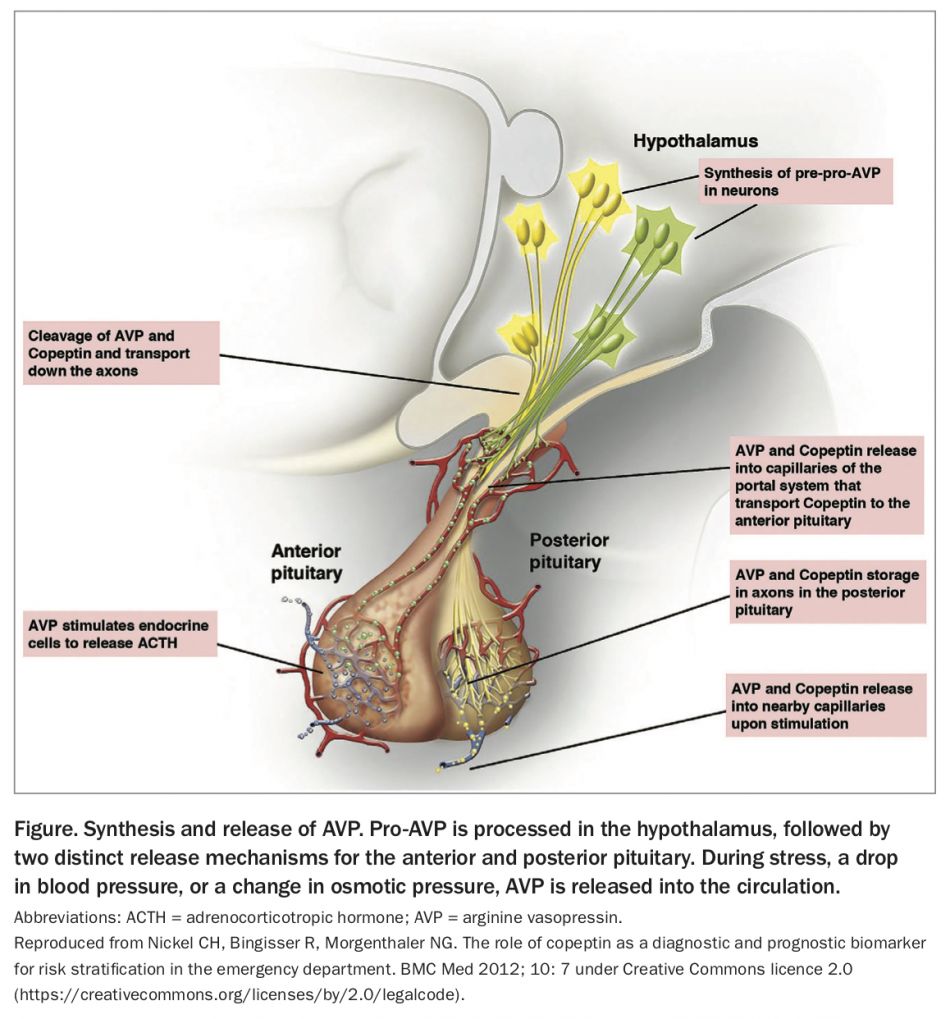{kind=link}
The principal physiological function of AVP is fluid homeostasis. Changes in extracellular fluid osmolality are detected by osmoreceptors, which project to magnocellular neurons to stimulate release of AVP into the posterior pituitary. The concentration of circulating AVP is directly related to serum osmolality, and a rise in plasma osmolality above about 280 mOsm/kg is the main stimulus for AVP and copeptin release. Above this threshold, a linear increase in AVP occurs with increasing osmolality.12
The actions of AVP are mediated by binding to one of three receptors (V1 to V3) on target cells.10 Regulation of water balance is mediated via AVP binding to V2 receptors on renal collecting tubular cells, leading to increased tubular fluid permeability via aquaporin-2 water channels, water retention and urinary concentration.10
AVP also has a role in regulation of the endocrine stress response. A second neurosecretory pathway transports high concentrations of AVP from parvocellular neurons to the pituitary portal system, where it has a neuroregulatory role in adrenocorticotrophin hormone (ACTH) release from the anterior pituitary.9 AVP and copeptin are also released in response to nonosmotic stimuli, including hypoglycaemia, sepsis, hypotension, exercise, and nausea and vomiting.11,13,14
Aetiology of polydipsia and polyuria
The aetiologies of polyuria-polydipsia syndrome include central DI, nephrogenic DI and primary polydipsia. The common causes of each are summarised in the Table.2
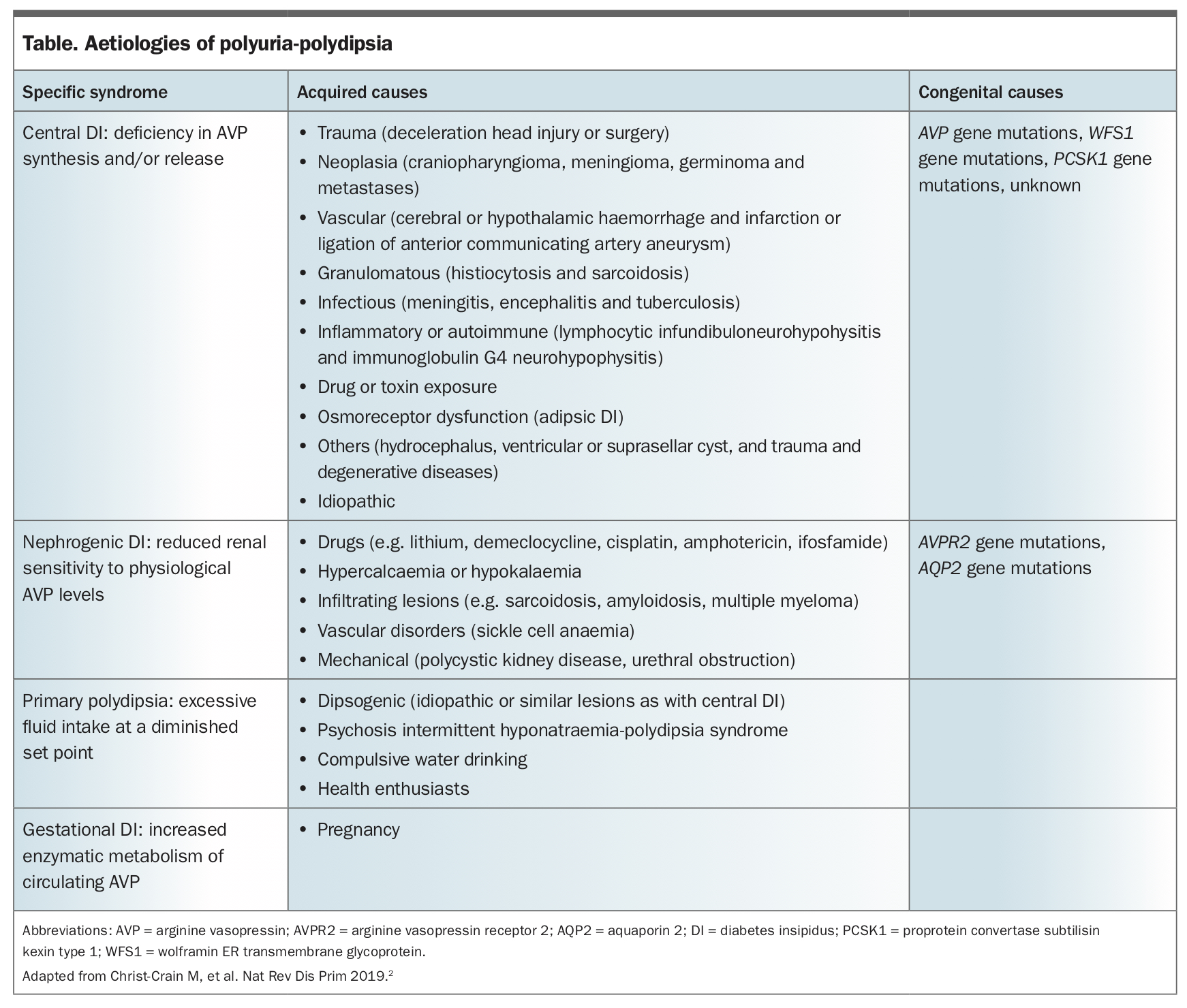{kind=link}
Diabetes insipidus
DI is rare, with a prevalence of 1 in 25,000 individuals.2 It can manifest at any age, with the age at presentation markedly dependent on aetiology. Acquired forms make up more than 90% of cases and present after early childhood, whereas hereditary forms contribute to less than 10% of cases and manifest early in life.15 Rarely, DI can present during pregnancy.2
Central DI
Central DI is the most common type of DI and is caused by deficiency of AVP production and/or secretion from AVP-producing magnocellular neurons. Clinical manifestations are variable, and range from mild to severe forms, depending on the extent of neuronal damage.2 Central DI can result from a variety of acquired and congenital causes. The most frequent presentation is following trans-sphenoidal surgery, with 16 to 34% of patients experiencing transient DI16-19 and 2 to 7% developing permanent DI.19,20 Other acquired causes include central nervous system tumours such as craniopharyngiomas, germ-cell tumours or metastases, central nervous system malformations, trauma, haemorrhage, and autoimmune and granulomatous disease. Primary pituitary tumours very rarely present with central DI, and the presence of DI in concert with a pituitary lesion should alert the physician to an alternative diagnosis.2 Congenital forms of DI are uncommon and include mutations in the AVP, WFS1 and PCSK1 genes.21
Nephrogenic DI
Nephrogenic DI occurs because of AVP resistance in the kidney;2 therefore, unlike in central DI, plasma AVP (and hence copeptin) is elevated in patients with this condition.22 Acquired aetiologies account for most cases, with lithium treatment being the most common cause. Up to 40% of people treated with lithium develop nephrogenic DI.23 Other drugs that possibly have the same effect include amphotericin, demeclocycline (only available in Australia through the Special Access Scheme), ifosfamide and cisplatin.24 Transient nephrogenic DI can occur in acute or chronic renal failure, hypokalaemia, hypercalcaemia and following obstructive nephropathy.24 Congenital nephrogenic DI is rare and most frequently results from mutations in the AVP-receptor 2 gene (AVPR2) (>90%), or less commonly, the aquaporin 2 (AQP2) gene (<10%).24,25 Most patients with congenital nephrogenic DI are male.25
Gestational DI
Gestational DI occurs due to accelerated degradation of AVP by increased activity of placental vasopressinase.26,27 Pre-existing asymptomatic partial central DI can also become symptomatic during pregnancy, owing to insufficient AVP secretion in response to increased degradation.28
Primary polydipsia
Primary polydipsia occurs from excessive fluid intake, which results in a decrease in serum osmolality, and consequently physiological inhibition of AVP, leading to polyuria.2 Primary polydipsia is most commonly associated with psychiatric conditions, including schizophrenia, anxiety disorders, depression and addictive disorders, or neurodevelopmental disorders, including autism and intellectual disability.29,30 It can also occur in people without these conditions, likely due to an abnormally low thirst threshold, a condition termed dipsogenic polydipsia.31 Primary polydipsia can result in severe hyponatraemia, particularly in individuals who participate in prolonged exercise such as marathon runners, where excessive intake of hypotonic fluids is paired with persistent ADH secretion.32
Investigation of polydipsia and polyuria
Once hypotonic polyuria (>3 L/24 h or >40–50 mL/kg/24 h) has been confirmed and causes of osmotic diuresis (such as hyperglycaemia or hypercalcaemia) excluded, the next step is to distinguish between central DI, nephrogenic DI and primary polydipsia (Flowchart). Determining the aetiology can be challenging. Milder, partial forms of central DI can be particularly difficult to distinguish from primary polydipsia.
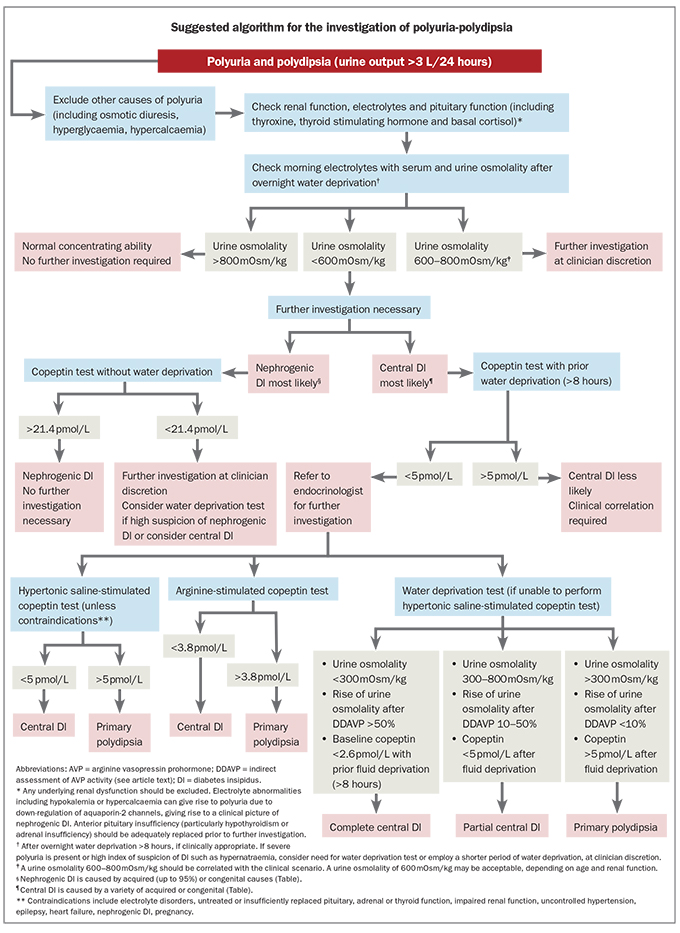{kind=link}
Clinical features such as headache and visual impairment should be sought along with medical history including any head trauma, psychiatric and dependency disorders, pregnancy, hypopituitarism, family history and a full medication list.1 Although clinical manifestations such as sudden onset of symptoms, preference for cold drinks, drinking at night and nocturia have been described as typical for DI, these symptoms are also reported by most patients with primary polydipsia.3 Similarly, while the incidence of psychiatric conditions has been reported as 27% in patients with primary polydipsia, they are also present in 17% of patients with central DI.3 Consequently, such clinical features are not reliable for distinguishing between these conditions.
Physical examination should include assessment of blood pressure, heart rate and fluid balance, as well as evaluation for anterior hypopituitarism, such as hypothyroidism, adrenal insufficiency and hypogonadism, and diplopia and visual field defects. Inflammatory, vascular, malignant or infectious processes may have systemic involvement, which could be detected through a comprehensive examination, particularly considering the respiratory, rheumatological, haematological and dermatological aspects, and the breasts.
Initial investigations
Initial investigations should include electrolyte and serum glucose levels, renal function tests, paired serum and urine osmolality measurements, thyroid function tests and basal early morning cortisol level.1 A lithium level should also be performed for patients taking lithium. Hypernatraemia and hyponatraemia may indicate DI or primary polydipsia, respectively, and may indicate a diagnosis without further investigations in the correct clinical context. However, many patients will have a plasma sodium level in the normal range.3 Urine osmolality greater than serum osmolality indicates water conservation is present. In patients with mild polyuria who can tolerate overnight water deprivation, a urine osmolality greater than 800 mOsm/kg indicates normal AVP function, and no further testing is required.1 All other patients should undergo further testing. In a patient with severe polyuria and/or high index of suspicion for DI, water deprivation, even for a few hours, may not be safe.
Basal copeptin measurements
Direct measurement of AVP is technically challenging. AVP has low plasma concentrations, a short half-life and is unstable in isolated plasma, which complicates sample preparation.4,5,33 Due to its small size, measuring AVP is also technically challenging and requires multiple extraction steps and analysis by competitive immunoassay, which is difficult to automate.7,8,34 AVP assays can require 1 mL or more of plasma or serum and generally take more than 48 hours to produce a result.9 Thus, measuring AVP has not been clinically useful. In comparison, copeptin, which is produced in equimolar amounts to AVP and correlates well with plasma AVP and serum osmolality, is easy to measure using automated chemiluminescence sandwich immunoassay.8,35,36 Copeptin assays can be performed on as little as 50 mcL of sample and can produce a result in less than two hours. Copeptin is highly stable ex vivo and samples can be stored at room temperature for several days.9 Two copeptin assays, Brahms KRYPTOR and Brahms LIA, have been used in the differential diagnosis of polyuria-polydipsia syndrome. Copeptin concentrations measured by the KRYPTOR and LIA assays have showed a good correlation over a wide range of copeptin concentrations.37
Unstimulated basal copeptin measurements are useful for the diagnosis of nephrogenic DI. In a patient with polyuria and polydipsia, a baseline copeptin without prior fluid deprivation of greater than 21.4 pmol/L appears to be diagnostic of partial or complete nephrogenic DI. However, studies to date have included small numbers of participants with nephrogenic DI and further data are needed for confirmation. A baseline plasma copeptin with prior water deprivation (>8 hours) of less than 2.6 pmol/L is highly suggestive of complete central DI;22 however, a stimulation test isusually needed to differentiate central DI from primary polydipsia.
Referral to endocrinologist
Patients with polyuria-polydipsia syndrome in whom other causes of osmotic diuresis have been excluded should be referred to an endocrinologist for consideration of dynamic testing to confirm the diagnosis. Dynamic testing should only be performed at experienced centres. Patients diagnosed with DI should be further investigated to elicit a cause and commenced on appropriate management under endocrinology supervision.
Water deprivation test
The traditional water deprivation test was the gold standard for the diagnosis of DI for many decades. This test is based on indirect assessment of AVP activity by measuring urine concentration capacity during prolonged fluid deprivation and then after administration of desmopressin (DDAVP), a synthetic AVP analogue.38 However, the water deprivation test has several limitations. Regardless of cause, chronic polyuria reduces the renal medullary concentration capacity and impairs the response to osmotic stimulation and exogenous vasopressin.39 Furthermore, many patients with partial central DI or nephrogenic DI retain a limited capacity to secrete or respond to AVP.38 Consequently, the overall diagnostic accuracy of the water deprivation test is only 70% and is particularly poor for primary polydipsia at 41%.40 The addition of copeptin to the traditional water deprivation test does not reliably improve the diagnostic accuracy, most likely due to thirsting alone being an insufficient osmotic stimulus for a significant increase in AVP and copeptin.3 The water deprivation test is burdensome and the test duration can be up to 18 hours, which has practical limitations.38 Furthermore, there is a risk of hyponatraemia with excessive fluid intake after administration of DDAVP, although this is not seen if aqueous argipressin, rather than DDAVP, is used in the test.
Hypertonic saline-stimulated copeptin test
In contrast to the challenges of the water deprivation test, intravenous infusion of 3% hypertonic saline produces a reliable osmotic stimulus within a couple of hours. The hypertonic saline test involves administration of 3% hypertonic saline until a target plasma sodium level of 150 mmol/L is reached, at which point blood is drawn for measurement of copeptin. The patient then receives water orally and 5% dextrose and the plasma sodium is rechecked in one hour to ensure normalisation before discharge. A hypertonic saline stimulated copeptin of greater than 4.9 pmol/L has a diagnostic accuracy of 96.5%, sensitivity of 93.2% and specificity of 100% for differentiating central DI, including milder forms, and primary polydipsia.3 Hypertonic saline-stimulated copeptin measurements have now replaced the water deprivation test as the gold standard test in investigation of polyuria-polydipsia. Limitations include the requirement for close monitoring, and adverse effects including transient hyperkalaemia and nausea and vomiting.3,41 Severe nausea and/or vomiting during the test can enhance the copeptin response, most likely through AVP and copeptin release from the parvocellular pathway, potentially causing false-negative results. The test should be interpreted with caution in these patients.42
Arginine-stimulated copeptin test
The amino acid arginine is an endogenous precursor of nitric oxide, which is an important signalling pathway of many endocrine regulatory pathways. Arginine infusion provides a nonosmotic stimulus for the release of AVP and copeptin and can therefore be used to help differentiate between central DI and primary polydipsia.43 A plasma copeptin test result of greater than 3.8 pmol/L 60 minutes after arginine infusion has a diagnostic accuracy of 93%, sensitivity of 93% and specificity of 92% for differentiating primary polydipsia from central DI.43 Although the diagnostic accuracy is slightly lower than the hypertonic saline test, the arginine test is quicker to perform and superior in terms of tolerability. Similarly to the hypertonic saline test, results should be interpreted with caution in patients who experienced severe nausea and/or vomiting during the test.43
Conclusion
Differentiating between central DI, nephrogenic DI and primary polydipsia is crucial in patients presenting with polyuria-polydipsia and in whom osmotic diuresis has been excluded. Nephrogenic DI can often be diagnosed with a basal plasma copeptin level of greater than 21.4 pmol/L without prior water deprivation. Central DI is suggested if basal plasma copeptin level is less than 2.6 pmol/L with prior water deprivation; however, stimulated copeptin measurements are generally required to differentiate between central DI and primary polydipsia, for which the hypertonic saline-stimulated copeptin test has now become the gold standard. ET
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.