Margaret, aged 78 years, is brought in by her family who are concerned about her malaise and mild confusion. A week ago, she was unwell with an influenza-like illness, with vomiting and diarrhoea that lasted for a couple of days. Although most of her symptoms have improved, she still feels weak. Her family have noticed that she is more forgetful than before and is unsteady on her feet. On examination, she is afebrile, her blood pressure is 140/85mmHg, her pulse is regular at 72bpm and her mucous membranes are moist. She is oriented to time and place but poorly attentive and slow to answer. Cardiorespiratory and abdominal examinations are unremarkable. She has no focal neurological signs. Urinalysis is negative for ketones, nitrites and leucocytes.
Margaret has a past history of dyslipidaemia, hypertension and type 2 diabetes. She takes atorvastatin 40mg, amlodipine 5mg and perindopril/indapamide 10mg/2.5mg daily. She also takes metformin 1000mg twice daily and gliclazide modified-release 30mg in the morning. She has been taking sertraline 100mg daily for depression since the death of her husband six months ago.
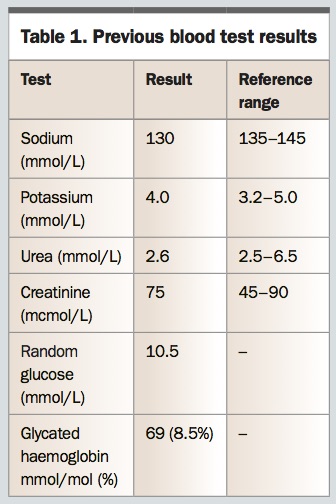
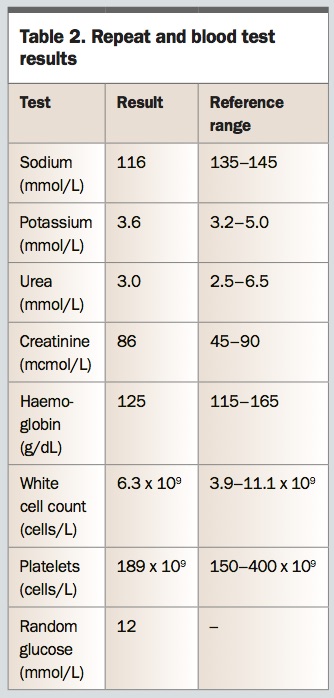
Margaret last attended the practice three months ago for her routine blood tests and prescriptions and was well at the time. You note the biochemistry results from that visit (Table 1) and order repeat and further tests (Table 2).
On assessing the results, you are concerned about the fall in serum sodium level in the past three months, along with the neurological changes, and you refer Margaret to the local emergency department.
What are the presenting signs and symptoms of hyponatraemia in the elderly?
Answer: The manifestations of hyponatraemia depend on both the severity and acuity of the low sodium concentration. Initial symptoms include nausea and malaise, but symptoms progress to headache, confusion, gait disturbance, seizures and coma as the sodium level falls. If hyponatraemia is acute, mild symptoms may develop at a sodium level of 130mmol/L or below, with risk of serious neurological compromise and death if it falls below 115 to 120mmol/L. Patients with chronic hyponatraemia may remain asymptomatic despite significantly low sodium levels and only present when the sodium level falls further, owing to an acute event. However, there is increasing recognition that chronic hyponatraemia may still be deleterious, despite the absence of obvious symptomatology. Hyponatraemia is associated with increased falls and fractures and is thought to directly affect bone resorption.1,2
Advertisement
Hyponatraemia is one of the most common electrolyte disorders in elderly patients, and age greater than 60 years is a risk factor. Clinical manifestations are more common and severe in the elderly and may be mistakenly attributed to other common conditions of ageing such as infection, cerebrovascular disease or dementia. A high degree of suspicion is warranted. A serum sodium test result should always be interpreted in the context of previous recent sodium levels and the rapidity of the change. When hyponatraemia is discovered in a patient new to a medical practice, a search for past pathology results is often valuable.
What conditions need to be excluded before a diagnosis of hyponatraemia is made?
Answer: Although the diagnosis of hyponatraemia is most likely in this clinical scenario, it is important to exclude the presence of ‘pseudohyponatraemia’, an artefactual lowering of measured sodium due to an increased solid component of plasma, typically proteins and lipids.
Sodium exists only in the aqueous phase of plasma. Reduction in the water component of plasma due to hypertriglyceridaemia or hyperproteinaemia will result in a spuriously low sodium result when the method of sodium analysis involves predilution of the sample. This is the most common technique used in high-throughput biochemistry laboratories. Blood gas analysers measure sodium directly without predilution and will reveal the true sodium concentration.
In the setting of pseudohyponatraemia, the measured osmolality of serum is normal, whereas calculated osmolarity (2 x Na + glucose + urea, in mmol/L), based on the erroneous sodium measurement, is low. This results in a raised ‘osmolar gap’ – the difference between measured osmolality and calculated osmolarity. When tests reveal hyponatraemia, measurement of serum osmolality is the next step. Normal measured osmolality with a raised osmolar gap should prompt measurement of lipids and proteins, including albumin and globulins. Marked elevation of triglycerides may occur in the setting of diabetes, pancreatitis, obstructive liver disease, nephrotic syndrome or familial syndromes. Raised globulins are a feature of the paraproteinaemias, including multiple myeloma, or may be found with therapeutic use of intravenous immunoglobulins.
How do glucose levels affect the assessment of sodium concentration?
Answer: Hyponatraemia may also be observed in the context of hyperglycaemia. Although sometimes grouped together with pseudohyponatraemia, the lowering of sodium in this context is real, not artefactual, due to the positive osmotic effect of glucose, which increases plasma volume. It is important to account for the prevailing glucose level when assessing and managing hyponatraemia, as the patient may not have true hypo-osmolar hyponatraemia and thus lack typical symptoms. Correction of the glucose level raises the sodium level concurrently and aggressive attempts to replace sodium are likely to result in overcorrection.
A ‘correction factor’ can be applied to determine the equivalent sodium level when glucose is normalised. Serum sodium decreases by 3mmol/L for every 10mmol/L increase in glucose concentration above normal. Thus, adjusted Na = measured Na + 0.3 x (glucose – 5.5) mmol/L.
Margaret’s measured plasma osmolality is 250mosmol/kg. Her calculated osmolarity is 2 x 116 + 12 + 3.0 = 247mmol/L. She has true hypo-osmolar hyponatraemia, consistent with her clinical presentation. Tests confirm that her triglyceride and protein levels are not markedly raised. Margaret’s sodium level, corrected for glucose, is 118mmol/L, which is not markedly different to the absolute sodium level but is still in the severely low range. She had a mildly low sodium level three months ago and is likely to have some degree of chronic hyponatraemia. In the absence of more recent testing, it is not possible to determine the acuity of the decrease in sodium level, but you suspect there has been a more rapid fall in the past week in relation to the intercurrent illness. You are therefore concerned that more serious neurological deficits may ensue if the sodium level is not promptly corrected.
Advertisement
What are the common causes of hypo-osmolar hyponatraemia in the elderly?
Answer: Sodium concentration depends on both the amount of sodium in the extracellular fluid, and the volume (water content) of the extracellular fluid. Hypo-osmolar hyponatraemia is generally classified into three categories based on volume status, as defined in
Box 1.
Hypovolaemic hyponatraemia occurs in the setting of sodium loss from the gastrointestinal tract, urinary tract or skin. Typical causes include vomiting, diarrhoea, salt-wasting nephropathies, diuretic use and mineralocorticoid deficiency (e.g. primary adrenal failure). The body’s response to hypovolaemia is to increase secretion of antidiuretic hormone (ADH, also known as vasopressin) from the posterior pituitary, which promotes water reabsorption in the kidney. As a result, there is relatively greater depletion of salt than water, leading to hyponatraemia.
Euvolaemic hyponatraemia typically results from excess ADH secretion, as seen in the syndrome of inappropriate antidiuretic hormone (SIADH). Causes of SIADH include medications (selective serotonin reuptake inhibitors, serotonin–noradrenaline reuptake inhibitors, NSAIDs, antiepileptics, benzodiazepines), stress (nausea, pain, surgery), secretory cancers (typically small-cell lung cancer) and other lung and CNS abnormalities. Hypothyroidism and glucocorticoid insufficiency can also impair renal water excretion and produce a similar picture. The kidneys are able to excrete excess free water up to a point, generally determined by solute load. On a regular diet, the kidneys can excrete up to 25L of water per day, depending on the glomerular filtration rate. When water consumption exceeds the kidney’s capacity to excrete it, hyponatraemia will result. This is seen in psychogenic polydipsia, a feature of some psychiatric conditions. It also occurs when renal water excretion is impaired due to a low glomerular filtration rate and/or low solute load in conditions such as anorexia and chronic alcoholism (when it is known as ‘beer potomania’), and in elderly women on a ‘tea and toast’ diet. Interestingly, individuals with diuretic-induced hyponatraemia may also present in a euvolaemic state. Pathogenic mechanisms are discussed below.
Hypervolaemic hyponatraemia occurs in conditions of fluid overload, such as acute or chronic renal failure, congestive cardiac failure, liver failure and nephrotic syndrome. Although there is increased total body water, this may be predominantly in the extravascular space. The decrease in effective arterial blood volume stimulates release of ADH, which further contributes to water retention and dilutional hyponatraemia.
What investigations distinguish potential causes of hyponatraemia?
Answer: Assessment of fluid status is the first step in differentiating causes of hyponatraemia. Examination of skin turgor, mucous membranes, orthostatic blood pressure and jugular venous pressure is useful, although this may be more challenging in the elderly because medications and other comorbidities may cloud assessment. Measuring plasma osmolality and urine sodium and osmolality are essential next steps (
Table 3). The normal physiological response to low sodium concentration is suppression of ADH secretion, which allows the kidney to excrete maximally dilute urine (urine osmolality <100mosmol/kg). Be aware of patients with pre-existing renal impairment in whom urine may not be fully dilute.
In hypovolaemic hyponatraemia, the urine sodium is typically low (less than 20mmol/L), as the renin–angiotensin–aldosterone system is activated to retain salt and water. The caveat is when salt and water losses are of renal origin, as occurs with continued diuretic use. When fluid loss is accompanied by metabolic alkalosis, such as in profuse vomiting, urine sodium may be greater than 20mmol/L, as sodium loss will accompany urinary bicarbonate loss. Urine osmolality often exceeds 450mosmol/kg as plasma volume depletion stimulates ADH secretion, appropriately increasing plasma volume but consequently diluting plasma sodium concentration.
In euvolaemic hyponatraemia due to SIADH, urine sodium is raised (typically greater than 40mmol/L), as is urine osmolality (greater than 100mosmol/kg). This is differentiated from primary polydipsia, where the mechanism for water excretion is intact and urine is maximally dilute (less than 100mosmol/kg).
Advertisement
In hypervolaemic hyponatraemia, urine sodium is usually low, unless there is concomitant diuretic use or renal dysfunction. Urine osmolality is high. The diagnosis is usually made clinically, owing to the obvious presence of oedema and a clinically apparent cardiac, hepatic or renal disorder.
The distinction between hypovolaemic and euvolaemic hyponatraemia can often be clinically challenging. Uric acid levels are typically elevated in patients who are dehydrated and reduced in those with SIADH and thiazide-induced hyponatraemia. Haematocrit and albumin level can also be helpful indicators of reduced plasma volume, increasing in the setting of hypovolaemia.
Thyroid stimulating hormone should be measured to exclude hypothyroidism as a cause of reduced water excretion. In rare cases, secondary hypothyroidism due to hypopituitarism may be a cause for hyponatraemia, so a free thyroxine (free T4) level should also be measured. A cortisol level taken before 9am is a simple screening test for glucocorticoid deficiency. It is difficult to specify an exact cortisol cut-off above which cortisol deficiency can be confidently excluded, but a level of around 450nmol/L has been suggested, depending on the local assay method and reference range. Levels of less than 150 nmol/L are highly suggestive of deficiency. If there is doubt about the diagnosis then a short synacthen test may be considered.
Margaret is assessed to be clinically euvolaemic. Her thyroid stimulating hormone and free T4 levels are normal (1.4mIU/L and 14pmol/L, respectively). A morning cortisol level is 483nmol/L. Her urine sodium is greater than 20nmol/L, but it is noted that she is currently taking perindopril/indapamide, an angiotensin-converting enzyme inhibitor/thiazide combination.
What factors could be contributing to Margaret’s presentation?
Answer: Thiazide diuretics increase urine sodium excretion. Angiotensin-converting enzyme inhibitors have been implicated in causing hyponatraemia, even without concomitant diuretic use, although this is much rarer. Clinically, there are multiple factors which may have contributed to Margaret’s presentation. Her use of sertraline, a selective serotonin reuptake inhibitor, may predispose towards SIADH and a mild underlying chronic hyponatraemia. The antihypertensive combination tablet would increase renal sodium losses and total body salt depletion and increase the risk of dehydration during illness, such as her recent bout of fever, vomiting and diarrhoea. In response to thirst, correction of dehydration with water rather than electrolyte-containing food and fluids can further compound the problem.
What are the features of thiazide-induced hyponatraemia?
Answer: Thiazide-induced hyponatraemia may occur within a few weeks of initiating treatment, or may occur after years of chronic use, if there are additional triggers. Predisposing factors for thiazide-induced hyponatraemia include female sex, increased age, low body weight, low salt intake, increased water intake and comorbidities such as type 2 diabetes, heart failure and chronic kidney disease. Acute lowering of plasma sodium concentrations can be precipitated by the addition of other medications affecting water homeostasis (
Box 2) and by a superimposed illness with gastrointestinal losses.
The risk of thiazide-induced hyponatraemia is dose-dependent and may be greater with hydrochlorothiazide compared with indapamide. Concurrent use of other medications that increase renal sodium loss, such as spironolactone and amiloride, compounds the risk. Thiazide-induced hyponatraemia may present as either euvolaemic or hypovolaemic hyponatraemia depending on the degree of concomitant ADH secretion, the underlying renal capacity to excrete free water, and salt and water intake. After thiazide cessation, it may take up to two weeks for the kidneys to recover and the serum sodium level to normalise.
Advertisement
What is the underlying pathophysiology of thiazide-induced hyponatraemia?
Answer: Thiazide diuretics block sodium chloride reabsorption in the distal tubule, hence impairing the kidney’s ability to excrete dilute urine. The resulting volume depletion stimulates ADH secretion via baroreceptors, leading to renal water retention and thirst-stimulated water intake. Thiazides also promote urinary potassium loss, increasing the risk of hypokalaemia. To compensate, intracellular potassium moves into the extracellular space in exchange for sodium, which further lowers serum sodium concentration. In the elderly, several physiological factors contribute to reduced water excretion, including low muscle mass, reduced protein intake, decreased renal prostaglandins and low glomerular filtration rate.3 Thus, older people are more prone to thiazide-related hyponatraemia. Female sex is also a risk factor. People with hypertension, heart disease or diabetes may also be following a low-salt diet on medical advice.
The precise factors that lead to thiazide-induced hyponatraemia in some individuals but not others remain poorly understood. Recently, a genome-wide association study demonstrated that a polymorphism in the gene encoding the prostaglandin transporter was found with double the frequency in people with thiazide-induced hyponatraemia versus control and general populations. Contrary to the conventional mechanistic understanding, people with thiazide-induced hyponatraemia had increased free water reabsorption but lower ADH levels, suggesting an underlying predisposition to water retention.4
Margaret is admitted to the medical high dependency unit, where her mental state, fluid balance and sodium levels can be closely monitored. Perindopril/indapamide is ceased and sertraline withheld for a few days until her sodium level starts to improve. Her salt intake is unrestricted and she is discouraged from excessive water consumption. Hormonal tests are not suggestive of hypothyroidism or glucocorticoid insufficiency. Margaret’s glucose levels improve with uptitration of the dose of gliclazide and closer attention to dietary measures.
How should hyponatraemia be corrected?
Answer: As a general rule, the speed of correction of hyponatraemia should take into account the rapidity of the fall in serum sodium level. Acute severe hyponatraemia developing over 48 hours or less generally manifests with neurological symptoms that can rapidly progress to seizures, coma and death. Immediate measures to raise the serum sodium level using hypertonic saline (3% NaCl) are usually required. Hypertonic saline can be given as repeated boluses or a continuous infusion. The most important factor in treatment success is frequent monitoring of serum sodium level to achieve specific targets within a set timeframe. An increase of 4 to 6mmol/L in serum sodium level in the first six hours is generally sufficient to improve neurological status. In situations where the decline in serum sodium level has been more gradual, and symptoms are less pronounced, slower correction is advised. Increases in serum sodium level should be steady and not exceed 8mmol/L in the first 24 hours and 18mmol/L in the first 48 hours. Levels should be corrected even more cautiously in the setting of longstanding hyponatraemia. When the acuity of hyponatraemia is not known, the more conservative approach to sodium correction is advised.
If isotonic or hypertonic fluids are being used to correct serum sodium level, infusions should be discontinued once the sodium level approaches the target level for the time period. If the level continues to rise above target, then redilution may be necessary, using 5% dextrose or, potentially, synthetic ADH (desmopressin) to avoid overcorrection.
Where thiazides are implicated in causing hyponatraemia, the offending agent should be ceased and, if necessary, replaced by alternative antihypertensives such as calcium channel blockers. As the thiazide effect wears off, restoration of water diuresis can still lead to rapid sodium correction, so close monitoring is recommended. In hypovolaemic hyponatraemia, cautious administration of normal saline is advised. Care needs to be taken in correction of potassium deficits. Administration of potassium-containing fluids can increase the risk of sodium overcorrection. In euvolaemic patients, fluid restriction may be helpful until the sodium level improves, as this encourages excretion of free water. As in Margaret’s case, hypertonic saline is generally not appropriate.
Advertisement
What are the potential complications of over-rapid correction?
Answer: Excessively rapid correction of serum sodium level results in the osmotic shift of water from the intracellular to extracellular space. This can lead to the phenomenon of osmotic demyelination, where the myelin sheaths surrounding axons are damaged. This most commonly occurs in the brainstem, especially the pontine region, where it manifests as dysarthria, dysphagia and flaccid paralysis. Neurological deficits may be irreversible. The risk of osmotic demyelination syndrome is higher in elderly women who take thiazides, and also in the setting of malnutrition, alcoholism and hypokalaemia, where it can occur even if recommended correction rates are adhered to.
How is chronic SIADH managed long term?
Answer: Where possible, the underlying cause of SIADH should be addressed. This might include cessation of offending drugs, treatment of cerebral or pulmonary infections, or surgery/chemotherapy for the underlying ADH-producing malignancy. In the setting of idiopathic SIADH, the mainstays of therapy are fluid restriction, oral salt therapy and urea. The latter two work by increasing the solute load and therefore allowing greater water excretion in the urine. In severe cases, oral solute loading can be combined with loop diuretics, which further dilute the urine. Lithium and demeclocycline (which is only available under the TGA Special Access Scheme) have been used off label to decrease the renal tubule responsiveness to ADH. However, their use is limited by risk of nephrotoxicity.
What is the role of the vaptans in the management of hyponatraemia?
Answer: Vasopressin receptor antagonists (‘vaptans’) block the binding of ADH to the V2 receptor in the collecting duct, thus preventing water reabsorption. This allows free water excretion, or ‘aquaresis’. Tolvaptan is an oral agent approved for use in Australia for the treatment of clinically significant hypervolaemic or euvolaemic hyponatraemia, including in patients with congestive heart failure and SIADH. It is not PBS listed, and its exact role in the management of these disorders is still under debate. It carries an increased risk of hepatotoxicity, should be initiated in a hospital setting and should not be used long term. The vaptans are not recommended in the management of hypovolaemic hyponatraemia or diuretic-induced hyponatraemia.5
Margaret’s serum sodium level slowly rises over the course of the week to above 130mmol/L and sertraline is restarted. You decide to avoid further use of thiazide diuretics. Her blood pressure remains stable but will require monitoring. She is discharged when her symptoms resolve and her neurological status returns to baseline. A month later, her serum sodium level remains borderline at 132mmol/L. You advise Margaret to avoid excessive fluid consumption, and, as her mood has stabilised, you both agree on a plan to slowly wean her antidepressant. (See Practice Points Box for advice on dealing with hyponatraemia). ET
COMPETING INTERESTS: None.
References
1. Usala RL, Fernandez SJ, Mete M, et al. Hyponatremia is associated with increased osteoporosis and bone fractures in a large US health system population. J Clin Endocrinol Metab 2015; 100: 3021-3031.
2. Verbalis JG, Barsony J, Sugimura Y, et al. Hyponatremia-induced osteoporosis. J Bone Miner Res 2010; 25: 554-563.
3. Liamis G, Filippatos TD, Elisaf MS. Thiazide-associated hyponatremia in the elderly: what the clinician needs to know. J Geriatr Cardiol 2016; 13: 175-182.
4. Ware JS, Wain LV, Channavajjhala SK, et al. Phenotypic and pharmacogenetic evaluation of patients with thiazide-induced hyponatremia. J Clin Invest 2017; 127: 3367-3374.
5. Woodward M Gonski P3, Grossmann M, Obeid J, Scholes R, Topliss DJ. Diagnosis and management of hyponatraemia in the older patient. Int Med J 2018; 48(Suppl 1): 5-12.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}